

Secuenciación de genomas y caracterización genética del virus de la influenza
 CompartirCompartir
CompartirCompartir
Secuenciación de genomas
Los virus de la influenza evolucionan constantemente; de hecho, todos los virus de la influenza sufren cambios genéticos con el tiempo (para obtener más información, consulte Cómo puede mutar el virus de la influenza: variaciones antigénicas menores y mayores). El genoma de un virus de la influenza consta de todos los genes que conforman el virus. Los CDC vigilan los virus de la influenza en circulación durante todo el año para monitorear los cambios que sufre el genoma (o partes del genoma) de estos virus. Esta tarea se realiza como parte de la vigilancia de rutina de la influenza en los Estados Unidos y de la función de los CDC como un centro que colabora con la Organización Mundial de la Salud (OMS) para referencia e investigación de la influenza. La información que los CDC recopilan del estudio de los cambios genéticos (también conocidos como "sustituciones", "variantes" o "mutaciones") que experimentan los virus de la influenza es importante para la salud pública porque ayuda a determinar si las vacunas existentes y las medidas paliativas médicas (por ej., los medicamentos antivirales) serán efectivas contra los nuevos virus de la influenza, además de determinar la posibilidad de que los virus de la influenza presentes en animales infecten a los seres humanos.



La secuenciación de genomas revela la secuencia de los nucleótidos en un gen, al igual que las letras del alfabeto en las palabras. La comparación de la composición de nucleótidos en el gen de un virus con el orden de los nucleótidos de otro gen puede demostrar ciertas variaciones entre los dos virus.
Las variaciones genéticas son importantes porque afectan la estructura de las proteínas de superficie del virus de la influenza. Las proteínas están formadas por secuencias de aminoácidos.
La sustitución de un aminoácido por otro puede afectar las características de un virus, como por ejemplo cuán bien se propaga un virus entre las personas y cuán susceptible es el virus a los medicamentos antivirales o a las vacunas actuales.
Los virus de la influenza A y B, los principales virus de la influenza que infectan a las personas, son virus del tipo RNA que cuentan con ocho segmentos de genes. Estos genes contienen "instrucciones" para fabricar virus nuevos; un virus de la influenza utiliza estas instrucciones después de infectar una célula humana para engañarla de modo que comience a fabricar más virus de la influenza y así diseminar la infección.
Los genes de la influenza constan de una secuencia de moléculas denominada nucleótidos que se unen entre sí y forman una cadena. Los nucleótidos se designan con las letras A, U, C o G.
La secuenciación de genomas es un proceso que determina el orden o la secuencia de los nucleótidos (por ej., A, U, C o G) en cada uno de los genes presentes en el genoma del virus. La secuenciación completa del genoma puede revelar la secuencia de alrededor de 13 500 letras de todos los genes del genoma del virus mientras que la secuenciación parcial del genoma muestra la secuencia de algunos o partes de esos genes.
Cada año, los CDC realizan la secuenciación completa del genoma de alrededor de 6 000 virus de la influenza de muestras clínicas originales tomadas a través de la vigilancia virológica. De los ochos genes que conforman un virus de la influenza A o B, los CDC se centran en la secuenciación de dos segmentos de genes: la hemaglutinina (HA) y la neuraminidasa (NA). Los genes de HA/NA determinan la estructura de las dos proteínas de superficie principales de los virus de la influenza. Las proteínas de superficie determinan las características importantes de los virus de la influenza, que incluyen la manera en que el virus de la influenza responde a los medicamentos antivirales, la similitud genética del virus con los virus de la vacuna contra la influenza y el potencial de los virus de la influenza zoonótica (origen animal) de infectar huéspedes humanos.
Caracterización genética
Los CDC y otros laboratorios de salud pública de todo el mundo han estado secuenciando los genes de los virus de la influenza desde la década de 1980. Actualmente, estas secuencias de genes están recopiladas en bases de datos para que puedan ser utilizadas por investigadores de salud pública como GenBank y la Iniciativa Global para Compartir Datos sobre la Gripe Aviar (GISAID). Las bibliotecas de secuencias de genes resultantes le permiten a los CDC y a otros laboratorios comparar los genes de los virus de la influenza actualmente en circulación con los genes de los virus de la influenza anteriores y los virus usados en las vacunas. A través de este proceso que compara las secuencias genéticas, denominado caracterización genética, los CDC pueden sacar suposiciones informadas sobre lo siguiente:
- Cómo los virus de la influenza se "relacionan" entre sí
- Cómo evolucionan los virus de la influenza
- Las variaciones genéticas (alias sustituciones o mutaciones) que aparecen cuando los virus comienzan a diseminarse más fácilmente, ocasionando enfermedades más graves o desarrollando resistencia a los medicamentos antivirales
- Cuán efectiva es la vacuna contra la influenza para brindar protección contra un virus de la influenza particular
- Adaptaciones en los virus de la influenza que circulan en las poblaciones animales que podrían permitir que el virus infecte a los seres humanos
Las diferencias relativas que se observan en un grupo de virus de la influenza se organizan y se muestran en un gráfico llamado "árbol filogenético". Los árboles filogenéticos para los virus de la influenza se asemejan a los árboles familiares (genealógicos) de las personas. Estos árboles muestran cuán estrecha es la relación entre cada uno de los virus. Los virus están agrupados según si los nucleótidos de sus genes son idénticos o no. Por lo general, los árboles filogenéticos de los virus de la influenza muestran la similitud que hay entre los genes de hemaglutinina (HA) o neuraminidasa (NA) de los virus. Cada secuencia de un virus de la influenza específico tiene asignada una rama del árbol. El grado de diferencia genética (cantidad de diferencias en los nucleótidos) entre los virus se representa con el largo de las líneas horizontales (ramas) en el árbol filogenético. Mientras más alejados están los virus en el eje horizontal del árbol filogenético, más diferentes genéticamente son unos de otros.
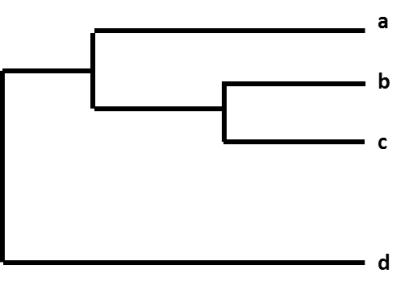
Figura. Árbol filogenético.
Por ejemplo, después de que los CDC establecen la secuencia de un virus de la influenza A(H3N2) recogido a través de la vigilancia, se cataloga la secuencia del virus teniendo en cuenta las secuencias de otros virus que tienen genes HA (H3) y NA (N2) similares. Como parte de este proceso, los CDC comparan la secuencia del nuevo virus con las secuencias de otros virus y observan las diferencias que hay entre ellos. Los CDC luego utilizan el árbol filogenético para representar visualmente la diferencia genética entre los virus H3N2.
Los CDC caracterizan genéticamente a los virus de la influenza todo el año. Estos datos genéticos se utilizan junto con los datos de la caracterización antigénica de los virus para determinar qué virus de la vacuna deberían elegirse para las próximas vacunas contra la influenza que se distribuirán en el Hemisferio Norte y Sur. Durante los meses posteriores a las reuniones de asesoramiento sobre la vacuna de la OMS, que se realizan en febrero y septiembre, los CDC recogen virus de la influenza a través de la vigilancia y compara las secuencias de los genes HA y NA de los virus de la vacuna actual con los de los virus de la influenza actualmente en circulación. Esta es una manera de evaluar cuán relacionados están los virus de la influenza en circulación con los virus contra los que debe brindar protección la vacuna contra la influenza estacional. Las diferencias pueden aparecer mientras se recogen los virus y se los caracteriza genéticamente.
Por ejemplo, a veces durante el transcurso de una temporada, los virus en circulación experimentan cambios genéticos, lo cual hace que se vuelvan totalmente diferentes de los virus de la vacuna correspondiente. Esto indica que quizás sea necesario seleccionar otro virus de la vacuna para la vacuna de la próxima temporada de influenza, a pesar de que otros factores, incluidos los resultados de la caracterización antigénica, influencian sobremanera las decisiones que se toman entorno a la vacuna. Las proteínas de superficie HA y NA de los virus de la influenza son antígenos, lo que significa que son reconocidos por el sistema inmunitario y además son capaces de desencadenar una respuesta inmunitaria, incluida la producción de anticuerpos que pueden detener la infección. La caracterización antigénica hace referencia al análisis de la reacción de un virus ante la presencia de anticuerpos para ayudar a evaluar cuán relacionado está con otro virus.
Métodos de secuenciación de genomas de la influenza
Una muestra de influenza contiene muchas partículas de virus de la influenza que crecieron en un tubo de ensayo y que, a menudo, presentan pequeñas diferencias genéticas entre sí en toda la población de virus semejantes.
Tradicionalmente, los científicos han utilizado la técnica de secuenciación denominada "el método de Sanger" para monitorear la evolución de la influenza como parte de la vigilancia virológica. La secuenciación de Sanger identifica la secuencia genética que predomina entre todos los virus de la influenza detectados en una muestra aislada. Esto quiere decir que las pequeñas variaciones en la población de virus presentes en una muestra no se ven reflejadas en el resultado final. A menudo, los científicos usan el método de Sanger para realizar la secuenciación parcial del genoma de los virus de la influenza pero existen tecnologías modernas (ver el próximo párrafo) que funcionan mejor para la secuenciación completa del genoma.
En los últimos cinco años, los CDC han utilizado las metodologías "secuenciación de próxima generación (NGS, por sus siglas en inglés), las cuales han ampliado significativamente la cantidad de información y detalles que pueden obtenerse del análisis de secuenciación. A diferencia de la secuenciación de Sanger, la NGS utiliza la detección molecular avanzada (AMD) para identificar las secuencias de genes de cada uno de los virus que componen una muestra. Por lo tanto, la NGS revela las variaciones genéticas que hay entre muchas partículas de virus de la influenza diferentes en una sola muestra; y estos métodos también ponen al descubierto toda la región de codificación de los genomas. Este nivel de detalle puede beneficiar directamente la toma de decisiones de la salud pública de maneras importantísimas aunque los datos deben ser cuidadosamente interpretados por expertos dentro del marco de otra información disponible. Ver Proyectos AMD: mejorar las vacunas contra la influenza para obtener más información sobre cómo la NGS y la AMD están revolucionando el esquema del genoma de la influenza en los CDC.
Recursos adicionales
Descargo de responsabilidad: Es posible que en este sitio encuentre algunos enlaces que le lleven a contenido disponible sólo en inglés. Además, el contenido que se ha traducido del inglés se actualiza a menudo, lo cual puede causar la aparición temporal de algunas partes en ese idioma hasta que se termine de traducir (generalmente en 24 horas). Llame al 1-800-CDC-INFO si tiene preguntas sobre la influenza estacional, cuyas respuestas no ha encontrado en este sitio. Agradecemos su paciencia.
Ayuda con los formatos de archivos:
¿Cómo se visualizan los diferentes formatos de archivos (PDF, DOC, PPT, MPEG) en este sitio?- Esta página fue revisada el 27 de sep. de 2017
- Esta página fue modificada el 27 de sep. de 2017
- Fuente del contenido:
- Centros para el Control y la Prevención de Enfermedades, Centro Nacional de Vacunación y Enfermedades Respiratorias (NCIRD, por sus siglas en inglés)
- Página mantenida por: Oficina del Director Adjunto de Comunicación, División de Noticias y Medios Electrónicos de Relaciones Públicas