Vasodilation
Vasodilation is the widening of blood vessels.[1] It results from relaxation of smooth muscle cells within the vessel walls, in particular in the large veins, large arteries, and smaller arterioles. The process is the opposite of vasoconstriction, which is the narrowing of blood vessels.
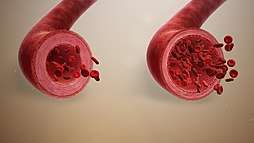
When blood vessels dilate, the flow of blood is increased due to a decrease in vascular resistance and increase in cardiac output. Therefore, dilation of arterial blood vessels (mainly the arterioles) decreases blood pressure. The response may be intrinsic (due to local processes in the surrounding tissue) or extrinsic (due to hormones or the nervous system). In addition, the response may be localized to a specific organ (depending on the metabolic needs of a particular tissue, as during strenuous exercise), or it may be systemic (seen throughout the entire systemic circulation).
Endogenous substances and drugs that cause vasodilation are termed vasodilators. Such vasoactivity is necessary for homeostasis (keeping the body running normally).
Function
The primary function of vasodilation is to increase blood flow in the body to tissues that need it most. This is often in response to a localized need for oxygen but can occur when the tissue in question is not receiving enough glucose, lipids, or other nutrients. Localized tissues have multiple ways to increase blood flow, including releasing vasodilators, primarily adenosine, into the local interstitial fluid, which diffuses to capillary beds, provoking local vasodilation.[2][3] Some physiologists have suggested that it is the lack of oxygen itself that causes capillary beds to vasodilate by the smooth muscle hypoxia of the vessels in the region. This latter hypothesis is posited due to the presence of precapillary sphincters in capillary beds. These approaches to the mechanism of vasodilation are not mutually exclusive.[4]
Vasodilation and arterial resistance
Vasodilation directly affects the relationship between mean arterial pressure, cardiac output, and total peripheral resistance (TPR). Vasodilation occurs in the time phase of cardiac systole, whereas vasoconstriction follows in the opposite time phase of cardiac diastole. Cardiac output (blood flow measured in volume per unit time) is computed by multiplying the heart rate (in beats per minute) and the stroke volume (the volume of blood ejected during ventricular systole). TPR depends on several factors, including the length of the vessel, the viscosity of blood (determined by hematocrit) and the diameter of the blood vessel. The latter is the most important variable in determining resistance, with the TPR changing by the fourth power of the radius. An increase in either of these physiological components (cardiac output or TPR) causes a rise in the mean arterial pressure. Vasodilation works to decrease TPR and blood pressure through relaxation of smooth muscle cells in the tunica media layer of large arteries and smaller arterioles.[5]
Vasodilation occurs in superficial blood vessels of warm-blooded animals when their ambient environment is hot; this process diverts the flow of heated blood to the skin of the animal, where heat can be more easily released to the atmosphere. The opposite physiological process is vasoconstriction. These processes are naturally modulated by local paracrine agents from endothelial cells (e.g., nitric oxide, bradykinin, potassium ions, and adenosine), as well as an organism's autonomic nervous system and adrenal glands, both of which secrete catecholamines such as norepinephrine and epinephrine, respectively.[6][7]
Examples and individual mechanisms
Vasodilation is the result of relaxation in smooth muscle surrounding the blood vessels. This relaxation, in turn, relies on removing the stimulus for contraction, which depends on intracellular calcium ion concentrations and is tightly linked with phosphorylation of the light chain of the contractile protein myosin. Thus, vasodilation works mainly either by lowering intracellular calcium concentration or by dephosphorylation (really substitution of ATP for ADP) of myosin. Dephosphorylation by myosin light-chain phosphatase and induction of calcium symporters and antiporters that pump calcium ions out of the intracellular compartment both contribute to smooth muscle cell relaxation and therefore vasodilation. This is accomplished through reuptake of ions into the sarcoplasmic reticulum via exchangers and expulsion across the plasma membrane.[8] There are three main intracellular stimuli that can result in the vasodilation of blood vessels. The specific mechanisms to accomplish these effects vary from vasodilator to vasodilator.
| Class | Description | Example |
|---|---|---|
| Hyperpolarization-mediated (Calcium channel blocker) | Changes in the resting membrane potential of the cell affects the level of intracellular calcium through modulation of voltage-sensitive calcium channels in the plasma membrane. | adenosine |
| cAMP-mediated | Adrenergic stimulation results in elevated levels of cAMP and protein kinase A, which results in increasing calcium removal from the cytoplasm. | prostacyclin |
| cGMP-mediated (Nitrovasodilator) | Through stimulation of protein kinase G. | nitric oxide |
PDE5 inhibitors and potassium channel openers can also have similar results.
Compounds that mediate the above mechanisms may be grouped as endogenous and exogenous.
Endogenous
| Vasodilators [9] | Receptor (↑ = opens. ↓ = closes) [9] On vascular smooth muscle cells if not otherwise specified |
Transduction (↑ = increases. ↓ = decreases) [9] |
|---|---|---|
| EDHF | ? | hyperpolarization → ↓VDCC → ↓intracellular Ca2+ |
PKG activity →
| ||
| NO receptor on endothelium | ↓endothelin synthesis [10] | |
| epinephrine (adrenaline) | β-2 adrenergic receptor | ↑Gs activity → ↑AC activity → ↑cAMP → ↑PKA activity → phosphorylation of MLCK → ↓MLCK activity → dephosphorylation of MLC |
| histamine | histamine H2 receptor | |
| prostacyclin | IP receptor | |
| prostaglandin D2 | DP receptor | |
| prostaglandin E2 | EP receptor | |
| VIP | VIP receptor | ↑Gs activity → ↑AC activity → ↑cAMP → ↑PKA activity →
|
| (extracellular) adenosine | A1, A2a and A2b adenosine receptors | ↑ATP-sensitive K+ channel → hyperpolarization → close VDCC → ↓intracellular Ca2+ |
| ↑P2Y receptor | activate Gq → ↑PLC activity → ↑intracellular Ca2+ → ↑NOS activity → ↑NO → (see nitric oxide) | |
| L-arginine | imidazoline and α-2 receptor? | Gi → ↓cAMP → activation of Na+/K+-ATPase[11] → ↓intracellular Na+ → ↑Na+/Ca2+ exchanger activity → ↓intracellular Ca2+ |
| bradykinin | bradykinin receptor | |
| substance P | ||
| niacin (as nicotinic acid only) | ||
| platelet-activating factor (PAF) | ||
| CO2 | - | ↓interstitial pH → ?[12] |
| interstitial lactic acid(probably) | - | |
| muscle work | - |
|
|
various receptors on endothelium | ↓endothelin synthesis [10] |
The vasodilating action of activation of beta-2 receptors (such as by adrenaline) appears to be endothelium-independent.[13]
Sympathetic nervous system vasodilation
Although it is recognized that the sympathetic nervous system plays an expendable role in vasodilation, it is only one of the mechanisms by which vasodilation can be accomplished. The spinal cord has both vasodilation and vasoconstriction nerves. The neurons that control vascular vasodilation originate in the hypothalamus. Some sympathetic stimulation of arterioles in skeletal muscle is mediated by epinephrine acting on β-adrenergic receptors of arteriolar smooth muscle, which would be mediated by cAMP pathways, as discussed above. However, it has been shown that knocking out this sympathetic stimulation plays little or no role in whether skeletal muscle is able to receive sufficient oxygen even at high levels of exertion, so it is believed that this particular method of vasodilation is of little importance to human physiology.[14]
In cases of emotional distress, this system may activate, resulting in fainting due to decreased blood pressure from vasodilation, which is referred to as vasovagal syncope.[15]
Cold-induced vasodilation
Cold-induced vasodilation (CIVD) occurs after cold exposure, possibly to reduce the risk of injury. It can take place in several locations in the human body but is observed most often in the extremities. The fingers are especially common because they are exposed most often.
When the fingers are exposed to cold, vasoconstriction occurs first to reduce heat loss, resulting in strong cooling of the fingers. Approximately five to ten minutes after the start of the cold exposure of the hand, the blood vessels in the finger tips will suddenly vasodilate. This is probably caused by a sudden decrease in the release of neurotransmitters from the sympathetic nerves to the muscular coat of the arteriovenous anastomoses due to local cold. The CIVD increases blood flow and subsequently the temperature of the fingers. This can be painful and is sometimes known as the 'hot aches' which can be painful enough to bring on vomiting.
A new phase of vasoconstriction follows the vasodilation, after which the process repeats itself. This is called the Hunting reaction. Experiments have shown that three other vascular responses to immersion of the finger in cold water are possible: a continuous state of vasoconstriction; slow, steady, and continuous rewarming; and a proportional control form in which the blood vessel diameter remains constant after an initial phase of vasoconstriction. However, the vast majority of responses can be classified as the Hunting reaction.[16]
Other mechanisms of vasodilation
Other suggested vasodilators or vasodilating factors include:
- absence of high levels of environmental noise
- absence of high levels of illumination
- adenosine - adenosine agonist, used primarily as an anti-arrhythmic
- alpha blockers (block the vasoconstricting effect of adrenaline)
- amyl nitrite and other nitrites are often used recreationally as a vasodilator, causing lightheadedness and a euphoric feeling
- atrial natriuretic peptide (ANP) - a weak vasodilator
- capsaicin (chili)[17]
- ethanol (alcohol)
- histamine-inducers
- Complement proteins C3a, C4a, and C5a work by triggering histamine release from mast cells and basophil granulocytes.
- nitric oxide inducers
- l-arginine (a key amino acid)
- glyceryl trinitrate (commonly known as nitroglycerin)
- isosorbide mononitrate and isosorbide dinitrate
- pentaerythritol tetranitrate (PETN)
- sodium nitroprusside
- PDE5 inhibitors: these agents indirectly increase the effects of nitric oxide
- sildenafil (Viagra)
- tadalafil (Cialis)
- vardenafil (Levitra)
- tetrahydrocannabinol (THC)
- theobromine
- minoxidil
- papaverine an alkaloid found in the opium poppy papaver somniferum
- estrogen
- apigenin: In rat small mesenteric arteries, apigenin acts on TRPV4 in endothelial cells to induce EDHF-mediated vascular dilation (Br J Pharmacol 2011 Nov 3)
Therapeutic uses
Vasodilators are used to treat conditions such as hypertension, wherein the patient has an abnormally high blood pressure, as well as angina, congestive heart failure, and erectile dysfunction, and where maintaining a lower blood pressure reduces the patient's risk of developing other cardiac problems.[5] Flushing may be a physiological response to vasodilators. Some phosphodiesterase inhibitors such as sildenafil, vardenafil and tadalafil, work to increase blood flow in the penis through vasodilation. They may also be used to treat pulmonary arterial hypertension (PAH).
Antihypertensives that work by opening blood vessels
- These drugs can keep vessels staying opened or help vessels refrain from being narrowed.[18]
- Drugs that appear to work by activating the α2A receptors in the brain thereby decreasing sympathetic nervous system activity.[19][18]
- According to American Heart Association, Alpha-methyldopa may cause Orthostatic syncope as it exerts a greater blood pressure lowering effect when one is standing upright which may lead to feeling weak or fainting if the blood pressure has been lowered too far. Methyldopa's prominent side effects include drowsiness or sluggishness, dryness of the mouth, fever or anemia. Additionally to these, male patients may experience impotence. [18]
- Clonidine, guanabenz or guanfacine may give rise to severe dryness of the mouth, constipation or drowsiness. Abrupt cessation taking may raise blood pressure quickly to dangerously high levels.[18]
- Directly relax the muscle in the walls of the blood vessels (especially the arterioles), allowing the vessel to dilate (widen). [18]
- Hydralazine may cause headaches, swelling around the eyes, heart palpitations or aches and pains in the joints. In clinical setting, hydralazine isn't usually used alone.[18]
- Minoxidil is a potent direct vasodilator used only in resistant severe high blood pressure or when kidney failure is present. Noted adverse effects comprise fluid retention (marked weight gain) and excessive hair growth.[18]
See also
- Arteriolar vasodilator
- Nitrophorin
- Vasodilatory shock
References
- "Definition of Vasodilation". MedicineNet.com. 27 April 2011. Archived from the original on 5 January 2012. Retrieved 13 January 2012.
- Costa, F; Biaggioni, I (May 1998). "Role of nitric oxide in adenosine-induced vasodilation in humans". Hypertension. 31 (5): 1061–4. doi:10.1161/01.HYP.31.5.1061. PMID 9576114.
- Sato A, Terata K, Miura H, Toyama K, Loberiza FR, Hatoum OA, Saito T, Sakuma I, Gutterman DD (April 2005). "Mechanism of vasodilation to adenosine in coronary arterioles from patients with heart disease". American Journal of Physiology. Heart and Circulatory Physiology. 288 (4): H1633–40. doi:10.1152/ajpheart.00575.2004. PMID 15772334.
- Guyton, Arthur; Hall, John (2006). "Chapter 17: Local and Humoral Control of Blood Flow by the Tissues". In Gruliow, Rebecca (ed.). Textbook of Medical Physiology (Book) (11th ed.). Philadelphia, Pennsylvania: Elsevier Inc. pp. 196–197. ISBN 978-0-7216-0240-0.
- Klablunde, Richard E. (29 April 2008). "Therapeutic Uses of Vasodilators". CVPharmacology. Archived from the original on 16 December 2008. Retrieved 3 December 2013.
- Charkoudian, Nisha (2010). "Mechanisms and modifiers of reflex induced cutaneous vasodilation and vasoconstriction in humans". Journal of Applied Physiology. American Physiological Society. 109 (4): 1221–1228. doi:10.1152/japplphysiol.00298.2010. ISSN 8750-7587. PMC 2963327. PMID 20448028.
- Johnson, John M.; Kellogg, Dean L. (2010). "Local thermal control of the human cutaneous circulation". Journal of Applied Physiology. American Physiological Society. 109 (4): 1229–1238. doi:10.1152/japplphysiol.00407.2010. ISSN 8750-7587. PMC 2963328. PMID 20522732.
- Webb, RC (December 2003). "Smooth muscle contraction and relaxation". Advances in Physiology Education. 27 (1–4): 201–6. doi:10.1152/advan.00025.2003. PMID 14627618.
- Unless else specified in box, then ref is: Walter F. Boron (2005). Medical Physiology: A Cellular And Molecular Approaoch. Elsevier/Saunders. ISBN 978-1-4160-2328-9. Page 479
- Rod Flower; Humphrey P. Rang; Maureen M. Dale; Ritter, James M. (2007). Rang & Dale's pharmacology. Edinburgh: Churchill Livingstone. ISBN 978-0-443-06911-6.
- Kurihara, Kinji; Nakanishi, Nobuo; Ueha, Takao (1 November 2000). "Regulation of Na+-K+-ATPase by cAMP-dependent protein kinase anchored on membrane via its anchoring protein". American Journal of Physiology. Cell Physiology. 279 (5): C1516–C1527. doi:10.1152/ajpcell.2000.279.5.c1516. PMID 11029299.
- Modin A, Björne H, Herulf M, Alving K, Weitzberg E, Lundberg JO (2001). "Nitrite-derived nitric oxide: a possible mediator of 'acidic-metabolic' vasodilation". Acta Physiol. Scand. 171 (1): 9–16. doi:10.1046/j.1365-201X.2001.00771.x. PMID 11350258.
- Schindler, C; Dobrev, D; Grossmann, M; Francke, K; Pittrow, D; Kirch, W (January 2004). "Mechanisms of beta-adrenergic receptor-mediated venodilation in humans". Clinical Pharmacology and Therapeutics. 75 (1): 49–59. doi:10.1016/j.clpt.2003.09.009. PMID 14749691.
- Guyton (2006) pp. 207-208
- Guyton (2006) p. 208
- Daanen, H. A. M. (2003). "Finger cold-induced vasodilation: a review". European Journal of Applied Physiology. 89 (5): 411–426. doi:10.1007/s00421-003-0818-2. PMID 12712346.
- Franco-Cereceda A, Rudehill A (August 1989). "Capsaicin-induced vasodilatation of human coronary arteries in vitro is mediated by calcitonin gene-related peptide rather than substance P or neurokinin A". Acta Physiologica Scandinavica. 136 (4): 575–80. doi:10.1111/j.1748-1716.1989.tb08704.x. PMID 2476911.
- "Types of Blood Pressure Medications". www.heart.org. 31 October 2017. Archived from the original on 8 January 2019. Retrieved 2 May 2019.
- "Guanfacine Monograph for Professionals". Drugs.com. American Society of Health-System Pharmacists. Retrieved 18 March 2019.
| Authority control |
|
|---|