Complement system
The complement system, also known as complement cascade, is a part of the immune system that enhances (complements) the ability of antibodies and phagocytic cells to clear microbes and damaged cells from an organism, promote inflammation, and attack the pathogen's cell membrane. It is part of the innate immune system,[1] which is not adaptable and does not change during an individual's lifetime. The complement system can, however, be recruited and brought into action by antibodies generated by the adaptive immune system.
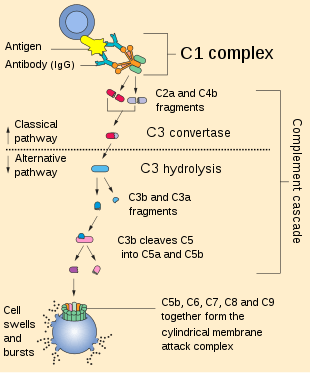
The complement system consists of a number of small proteins that are synthesized by the liver, and circulate in the blood as inactive precursors. When stimulated by one of several triggers, proteases in the system cleave specific proteins to release cytokines and initiate an amplifying cascade of further cleavages. The end result of this complement activation or complement fixation cascade is stimulation of phagocytes to clear foreign and damaged material, inflammation to attract additional phagocytes, and activation of the cell-killing membrane attack complex. Over 30 proteins and protein fragments make up the complement system, including serum proteins, and cell membrane receptors. They account for about 10% of the globulin fraction of blood serum.
Three biochemical pathways activate the complement system: the classical complement pathway, the alternative complement pathway, and the lectin pathway.[2]
History
In 1888, George Nuttall found that sheep blood serum had mild killing activity against the bacterium that causes anthrax. The killing activity disappeared when he heated the blood.[3] In 1891, Hans Ernst August Buchner, noting the same property of blood in his experiments, named the killing property "alexin", which means "to ward off" in Greek.[4] By 1894, several laboratories had demonstrated that serum from guinea pigs that had recovered from cholera killed the cholera bacterium in vitro. Heating the serum destroyed its killing activity. Nevertheless, the heat-inactivated serum, when injected into guinea pigs exposed to the cholera bacteria, maintained its ability to protect the animals from illness. Jules Bordet, a young Belgian scientist in Paris at the Pasteur Institute, concluded that this principle has two components, one that maintained a "sensitizing" effect after being heated and one (alexin) whose toxic effect was lost after being heated. The heat-stable component was responsible for immunity against specific microorganisms, whereas the heat-sensitive component was responsible for the non-specific antimicrobial activity conferred by all normal sera. In 1899, Paul Ehrlich renamed the heat-sensitive component "complement."[3]
Ehrlich introduced the term "complement" as part of his larger theory of the immune system. According to this theory, the immune system consists of cells that have specific receptors on their surface to recognize antigens. Upon immunisation with an antigen, more of these receptors are formed, and they are then shed from the cells to circulate in the blood. Those receptors, which we now call "antibodies", were called by Ehrlich "amboceptors" to emphasise their bifunctional binding capacity: They recognise and bind to a specific antigen, but they also recognise and bind to the heat-labile antimicrobial component of fresh serum. Ehrlich, therefore, named this heat-labile component "complement", because it is something in the blood that "complements" the cells of the immune system. Ehrlich believed that each antigen-specific amboceptor has its own specific complement, whereas Bordet believed that there is only one type of complement. In the early 20th century, this controversy was resolved when it became understood that complement can act in combination with specific antibodies, or on its own in a non-specific way.
Functions
.png)
Complement triggers the following immune functions:[5]
- Phagocytosis – by opsonizing antigens. C3b has most important opsonizing activity
- Inflammation – by attracting macrophages and neutrophils
- Membrane attack – by rupturing cell wall of bacteria
Overview
Most of the proteins and glycoproteins that constitute the complement system are synthesized by hepatocytes. But significant amounts are also produced by tissue macrophages, blood monocytes, and epithelial cells of the genitourinary system and gastrointestinal tract. The three pathways of activation all generate homologous variants of the protease C3-convertase. The classical complement pathway typically requires antigen-antibody complexes for activation (specific immune response), whereas the alternative pathway can be activated by spontaneous complement component 3 (C3) hydrolysis, foreign material, pathogens, or damaged cells. The mannose-binding lectin pathway can be activated by C3 hydrolysis or antigens without the presence of antibodies (non-specific immune response). In all three pathways, C3-convertase cleaves and activates component C3, creating C3a and C3b, and causes a cascade of further cleavage and activation events. C3b binds to the surface of pathogens, leading to greater internalization by phagocytic cells by opsonization.
In the alternative pathway, C3b binds to Factor B. Factor D releases Factor Ba from Factor B bound to C3b. The complex of C3b(2)Bb is a protease which cleaves C5 into C5b and C5a. C5 convertase is also formed by the classical pathway when C3b binds C4b and C2b. C5a is an important chemotactic protein, helping recruit inflammatory cells. C3a is the precursor of an important cytokine (adipokine) named ASP (although this is not universally accepted [6]) and is usually rapidly cleaved by carboxypeptidase B. Both C3a and C5a have anaphylatoxin activity, directly triggering degranulation of mast cells as well as increasing vascular permeability and smooth muscle contraction.[6] C5b initiates the membrane attack pathway, which results in the membrane attack complex (MAC), consisting of C5b, C6, C7, C8, and polymeric C9.[7] MAC is the cytolytic endproduct of the complement cascade; it forms a transmembrane channel, which causes osmotic lysis of the target cell. Kupffer cells and other macrophage cell types help clear complement-coated pathogens. As part of the innate immune system, elements of the complement cascade can be found in species earlier than vertebrates; most recently in the protostome horseshoe crab species, putting the origins of the system back further than was previously thought.
Classical pathway
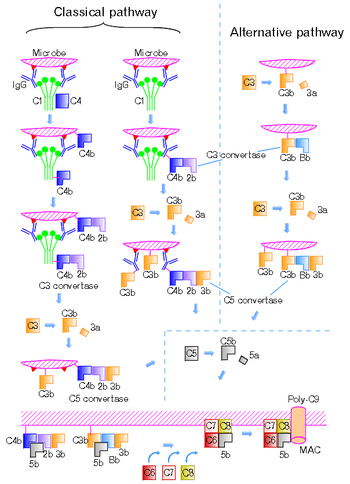
The classical pathway is triggered by activation of the C1-complex. The C1-complex is composed of 1 molecule of C1q, 2 molecules of C1r and 2 molecules of C1s, or C1qr2s2. This occurs when C1q binds to IgM or IgG complexed with antigens. A single pentameric IgM can initiate the pathway, while several, ideally six, IgGs are needed. This also occurs when C1q binds directly to the surface of the pathogen. Such binding leads to conformational changes in the C1q molecule, which leads to the activation of two C1r molecules. C1r is a serine protease. They then cleave C1s (another serine protease). The C1r2s2 component now splits C4 and then C2, producing C4a, C4b, C2a, and C2b (historically, the larger fragment of C2 was called C2a but is now referred to as C2b). C4b and C2b bind to form the classical pathway C3-convertase (C4b2b complex), which promotes cleavage of C3 into C3a and C3b. C3b later joins with C4b2b to make C5 convertase (C4b2b3b complex).
Alternative pathway
The alternative pathway is continuously activated at a low level, analogous to a car engine at idle, as a result of spontaneous C3 hydrolysis due to the breakdown of the internal thioester bond (C3 is mildly unstable in aqueous environment). The alternative pathway does not rely on pathogen-binding antibodies like the other pathways.[2] C3b that is generated from C3 by a C3 convertase enzyme complex in the fluid phase is rapidly inactivated by factor H and factor I, as is the C3b-like C3 that is the product of spontaneous cleavage of the internal thioester. In contrast, when the internal thioester of C3 reacts with a hydroxyl or amino group of a molecule on the surface of a cell or pathogen, the C3b that is now covalently bound to the surface is protected from factor H-mediated inactivation. The surface-bound C3b may now bind factor B to form C3bB. This complex in the presence of factor D will be cleaved into Ba and Bb. Bb will remain associated with C3b to form C3bBb, which is the alternative pathway C3 convertase.
The C3bBb complex is stabilized by binding oligomers of factor P (properdin). The stabilized C3 convertase, C3bBbP, then acts enzymatically to cleave much more C3, some of which becomes covalently attached to the same surface as C3b. This newly bound C3b recruits more B, D and P activity and greatly amplifies the complement activation. When complement is activated on a cell surface, the activation is limited by endogenous complement regulatory proteins, which include CD35, CD46, CD55 and CD59, depending on the cell. Pathogens, in general, don't have complement regulatory proteins (there are many exceptions, which reflect adaptation of microbial pathogens to vertebrate immune defenses). Thus, the alternative complement pathway is able to distinguish self from non-self on the basis of the surface expression of complement regulatory proteins. Host cells don't accumulate cell surface C3b (and the proteolytic fragment of C3b called iC3b) because this is prevented by the complement regulatory proteins, while foreign cells, pathogens and abnormal surfaces may be heavily decorated with C3b and iC3b. Accordingly, the alternative complement pathway is one element of innate immunity.
Once the alternative C3 convertase enzyme is formed on a pathogen or cell surface, it may bind covalently another C3b, to form C3bBbC3bP, the C5 convertase. This enzyme then cleaves C5 to C5a, a potent anaphylatoxin, and C5b. The C5b then recruits and assembles C6, C7, C8 and multiple C9 molecules to assemble the membrane attack complex. This creates a hole or pore in the membrane that can kill or damage the pathogen or cell.
Lectin pathway
The lectin pathway is homologous to the classical pathway, but with the opsonin, mannose-binding lectin (MBL), and ficolins, instead of C1q. This pathway is activated by binding of MBL to mannose residues on the pathogen surface, which activates the MBL-associated serine proteases, MASP-1, and MASP-2 (very similar to C1r and C1s, respectively), which can then split C4 into C4a and C4b and C2 into C2a and C2b. C4b and C2b then bind together to form the classical C3-convertase, as in the classical pathway. Ficolins are homologous to MBL and function via MASP in a similar way. Several single-nucleotide polymorphisms have been described in M-ficolin in humans, with effect on ligand-binding ability and serum levels. Historically, the larger fragment of C2 was named C2a, but it is now referred to as C2b.[8] In invertebrates without an adaptive immune system, ficolins are expanded and their binding specificities diversified to compensate for the lack of pathogen-specific recognition molecules.
Complement protein fragment nomenclature
Immunology textbooks have used different naming assignments for the smaller and larger fragments of C2 as C2a and C2b. The preferred assignment appears to be that the smaller fragment be designated as C2a: as early as 1994, a well known textbook recommended that the larger fragment of C2 should be designated C2b.[9] However, this was amplified in their 1999 4th edition, to say that:[10] "It is also useful to be aware that the larger active fragment of C2 was originally designated C2a, and is still called that in some texts and research papers. Here, for consistency, we shall call all large fragments of complement b, so the larger fragment of C2 will be designated C2b. In the classical and lectin pathways the C3 convertase enzyme is formed from membrane-bound C4b with C2b."[10]
This nomenclature is used in another literature:[11] "(Note that, in older texts, the smaller fragment is often called C2b, and the larger one is called C2a for historical reason.)"[12] The assignment is mixed in the latter literature, though. Some sources designate the larger and smaller fragments as C2a and C2b respectively[13][14][15][16][17][18][19][20][21] while other sources apply the converse.[9][10][22][23][24] However, due to the widely established convention, C2b here is the larger fragment, which, in the classical pathway, forms C4b2b (classically C4b2a). It may be noteworthy that, in a series of editions of Janeway's book, 1st to 7th, in the latest edition[20] they withdraw the stance to indicate the larger fragment of C2 as C2b.
Viral inhibition
Fixation of the MBL protein on viral surfaces has also been shown to enhance neutralization of viral pathogens.[25]
Review
| Activation pathway | Classic | Alternative | Lectin |
|---|---|---|---|
| Activator | Ag–Ab Complex | spontaneous hydrolysis of C3 | MBL-Mannose Complex |
| C3-convertase | C4b2a | C3bBb | C4b2a |
| C5-convertase | C4b2a3b | C3bC3bBb | C4b2a3b |
| MAC development | C5b+C6+C7+C8+C9 | ||
Activation of complements by antigen-associated antibody
In the classical pathway, C1 binds with its C1q subunits to Fc fragments (made of CH2 region) of IgG or IgM, which has formed a complex with antigens. C4b and C3b are also able to bind to antigen-associated IgG or IgM, to its Fc portion.[11][17][20]
Such immunoglobulin-mediated binding of the complement may be interpreted as that the complement uses the ability of the immunoglobulin to detect and bind to non-self antigens as its guiding stick. The complement itself can bind non-self pathogens after detecting their pathogen-associated molecular patterns (PAMPs),[20] however, utilizing specificity of the antibody, complements can detect non-self enemies much more specifically.
Some components have a variety of binding sites. In the classical pathway, C4 binds to Ig-associated C1q and C1r2s2 enzyme cleaves C4 to C4b and 4a. C4b binds to C1q, antigen-associated Ig (specifically to its Fc portion), and even to the microbe surface. C3b binds to antigen-associated Ig and to the microbe surface. Ability of C3b to bind to antigen-associated Ig would work effectively against antigen-antibody complexes to make them soluble.
Regulation
The complement system has the potential to be extremely damaging to host tissues, meaning its activation must be tightly regulated. The complement system is regulated by complement control proteins, which are present at a higher concentration in the blood plasma than the complement proteins themselves. Some complement control proteins are present on the membranes of self-cells preventing them from being targeted by complement. One example is CD59, also known as protectin, which inhibits C9 polymerisation during the formation of the membrane attack complex. The classical pathway is inhibited by C1-inhibitor, which binds to C1 to prevent its activation.
C3-convertase can be inhibited by Decay accelerating factor (DAF), which is bound to erythrocyte plasma membranes via a GPI anchor.
Role in disease
Complement deficiency
It is thought that the complement system might play a role in many diseases with an immune component, such as Barraquer–Simons Syndrome, asthma, lupus erythematosus, glomerulonephritis, various forms of arthritis, autoimmune heart disease, multiple sclerosis, inflammatory bowel disease, paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome and ischemia-reperfusion injuries,[26][27] and rejection of transplanted organs.[28]
The complement system is also becoming increasingly implicated in diseases of the central nervous system such as Alzheimer's disease and other neurodegenerative conditions such as spinal cord injuries.[29][30][31]
Deficiencies of the terminal pathway predispose to both autoimmune disease and infections (particularly Neisseria meningitidis, due to the role that the membrane attack complex ("MAC") plays in attacking Gram-negative bacteria).[32]
Infections with N. meningitidis and N. gonorrhoeae are the only conditions known to be associated with deficiencies in the MAC components of complement.[33] 40–50% of those with MAC deficiencies experience recurrent infections with N. meningitidis.[34]
Deficiencies in complement regulators
Mutations in the complement regulators factor H and membrane cofactor protein have been associated with atypical hemolytic uremic syndrome.[35][36] Moreover, a common single nucleotide polymorphism in factor H (Y402H) has been associated with the common eye disease age-related macular degeneration.[37] Polymorphisms of complement component 3, complement factor B, and complement factor I, as well as deletion of complement factor H-related 3 and complement factor H-related 1 also affect a person's risk of developing age-related macular degeneration.[38] Both of these disorders are currently thought to be due to aberrant complement activation on the surface of host cells.
Mutations in the C1 inhibitor gene can cause hereditary angioedema, a genetic condition resulting from reduced regulation of bradykinin by C1-INH.
Paroxysmal nocturnal hemoglobinuria is caused by complement breakdown of RBCs due to an inability to make GPI. Thus the RBCs are not protected by GPI anchored proteins such as DAF.[39]
Diagnostic tools
Diagnostic tools to measure complement activity include the total complement activity test.[40]
The presence or absence of complement fixation upon a challenge can indicate whether particular antigens or antibodies are present in the blood. This is the principle of the complement fixation test.
Modulation by infections
Recent research has suggested that the complement system is manipulated during HIV/AIDS, in a way that further damages the body.[41]
References
- Janeway, CA Jr; Travers P; Walport M; et al. (2001). "The complement system and innate immunity". Immunobiology: The Immune System in Health and Disease. New York: Garland Science. Retrieved 25 February 2013.
- Abbas AK, Lichtman AH, Pillai S (2010). Cellular and Molecular Immunology (6th ed.). Elsevier. pp. 272–288. ISBN 978-1-4160-3123-9.
- Chaplin H (2005). "Review: the burgeoning history of the complement system 1888–2005". Immunohematology / American Red Cross. 21 (3): 85–93. PMID 16178664.
- Nesargikar PN, Spiller B, Chavez R (2012). "The complement system: history, pathways, cascade and inhibitors". European Journal of Microbiology & Immunology. 2 (2): 103–11. doi:10.1556/EuJMI.2.2012.2.2. PMC 3956958. PMID 24672678.
- Murphy, Kenneth; Weaver, Casey (2017). "Innate Immunity: the First Lines of Defense". Janeway's Immunobiology (9th ed.). Garland Science. p. 49. ISBN 978-0-8153-4505-3.
- Klos, A.; Wende, E.; Wareham, K. J.; Monk, P. N. (2013). "International Union of Pharmacology. LXXXVII. Complement Peptide C5a, C4a, and C3a Receptors". Pharmacological Reviews. 65 (1): 500–43. doi:10.1124/pr.111.005223. PMID 23383423.
- Goldman AS, Prabhakar BS (1996). "The Complement System". In Baron S, et al. (eds.). Baron's Medical Microbiology (4th ed.). Univ of Texas Medical Branch. ISBN 978-0-9631172-1-2.
- Ammitzbøll, Christian Gytz; Kjær, Troels Rønn; Steffensen, Rudi; Stengaard-Pedersen, Kristian; Nielsen, Hans Jørgen; Thiel, Steffen; Bøgsted, Martin; Jensenius, Jens Christian (2012). "Non-Synonymous Polymorphisms in the FCN1 Gene Determine Ligand-Binding Ability and Serum Levels of M-Ficolin". PLoS ONE. 7 (11): e50585. Bibcode:2012PLoSO...750585A. doi:10.1371/journal.pone.0050585. PMC 3509001. PMID 23209787.
- Janeway C, Travers P (1994). Immunobiology : The Immune System in Health and Disease. London; San Francisco; New York: Current Biology Limited; Garland Pub. Inc., ISBN 0-8153-1691-7.
- Janeway CA, Travers P, Walport M, Capra JD (1999). Immunobiology: The Immune System in Health and Disease (4th ed.), 635p. New York: Garland Pub, ISBN 0-8153-3217-3.
- Abbas AK, Lichtman AH (2003). Cellular and Molecular Immunology (5th ed.), 563p. Philadelphia: Saunders, ISBN 0-7216-0008-5.
- p. 332
- Peakman M, Vergani D (1997). Basic and Clinical Immunology. New York: Churchill Livingstone, ISBN 0-443-04672-7.
- Paul WE (ed.) (1999). Fundamental Immunology (4th ed.), 1589p. Philadelphia: Lippincott-Raven, ISBN 0-7817-1412-5.
- Sims PJ, Wiedmer T (2000). "Complement biology," In Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, McGlave P, (eds.) (2000). Hematology: Basic Principles and Practice, 3rd ed. pp. 651–667, New York; Edinburgh: Churchill-Livingstone, ISBN 0-443-07954-4.
- Frank K, Atkinson JP (2001) Complement system. In Austen KF, Frank K, Atkinson JP, Cantor H. (ed.) Samter's Immunologic Diseases (6th ed.) Vol. 1, p. 281–298, Philadelphia: Lippincott Williams & Wilkins, ISBN 0-7817-2120-2.
- Roitt I, Brostoff J, Male D (2001). Immunology (6th ed.), 480p. St. Louis: Mosby, ISBN 0-7234-3189-2.
- Anderson DM (2003) Dorland's Illustrated Medical Dictionary (30th ed.), Philadelphia: W.B. Saunders, ISBN 0-7216-0146-4.
- Parham P (2005). The Immune System. New York: Garland, ISBN 0-8153-4093-1.
- Murphy K, Travers P, Walport M, with contributions by Ehrenstein M et al. (2008). Janeway's Immunobiology (7th ed.), New York: Garland Science, ISBN 0-8153-4123-7.
- Atkinson JP (2009). "Complement system," In Firestein GS, Budd RC, Harris ED Jr, McInnes IB, Ruddy S, Sergent JS, eds. (2009). Kelley's Textbook of Rheumatology, pp. 323–336, Philadelphia, PA: Saunders/Elsevier, ISBN 978-1-4160-3285-4.
- Janeway CA Jr.; Travers P; Walport M; Shlomchik MJ (2001). "The complement system and innate immunity". Immunobiology (5th ed.). Garland Publishing. ISBN 978-0-8153-3642-6.
- Doan T, Melvold R, Viselli S, Waltenbaugh C (2007). Lippincott's Illustrated Reviews: Immunology, 320p. Lippincott Williams & Wilkins
- DeFranco AL, Locksley RM, Robertson M (2007). Immunity : The Immune Response in Infectious and Inflammatory Disease. London; Sunderland, MA: New Science Press; Sinauer Associates, ISBN 978-0-9539181-0-2.
- Stoermer, Kristina A.; Morrison, Thomas E. (15 March 2011). "Complement and Viral Pathogenesis". Virology. 411 (2): 362–373 Central. doi:10.1016/j.virol.2010.12.045. PMC 3073741. PMID 21292294.
- Arumugam, Thiruma V; Shiels, Ian A; Woodruff, Trent M; Granger, D Neil; Taylor, Stephen M (2004). "The Role of the Complement System in Ischemia-Reperfusion Injury". Shock. 21 (5): 401–9. doi:10.1097/00024382-200405000-00002. PMID 15087815.
- Naesens, M.; Li, L.; Ying, L.; Sansanwal, P.; Sigdel, T. K.; Hsieh, S.-C.; Kambham, N.; Lerut, E.; Salvatierra, O.; Butte, A. J.; Sarwal, M. M. (2009). "Expression of Complement Components Differs Between Kidney Allografts from Living and Deceased Donors". Journal of the American Society of Nephrology. 20 (8): 1839–51. doi:10.1681/ASN.2008111145. PMC 2723986. PMID 19443638.
- Sacks, Steven H; Chowdhury, Paramit; Zhou, Wuding (2003). "Role of the complement system in rejection". Current Opinion in Immunology. 15 (5): 487–92. doi:10.1016/S0952-7915(03)00100-6. PMID 14499254.
- Galvan, M. D.; Luchetti, S.; Burgos, A. M.; Nguyen, H. X.; Hooshmand, M. J.; Hamers, F. P. T.; Anderson, A. J. (2008). "Deficiency in Complement C1q Improves Histological and Functional Locomotor Outcome after Spinal Cord Injury". Journal of Neuroscience. 28 (51): 13876–88. doi:10.1523/JNEUROSCI.2823-08.2008. PMC 2680920. PMID 19091977.
- Nguyen, Hal X; Galvan, Manuel D; Anderson, Aileen J (2008). "Characterization of early and terminal complement proteins associated with polymorphonuclear leukocytes in vitro and in vivo after spinal cord injury". Journal of Neuroinflammation. 5: 26. doi:10.1186/1742-2094-5-26. PMC 2443364. PMID 18578885.
- Beck, K. D.; Nguyen, H. X.; Galvan, M. D.; Salazar, D. L.; Woodruff, T. M.; Anderson, A. J. (2010). "Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment". Brain. 133 (2): 433–47. doi:10.1093/brain/awp322. PMC 2858013. PMID 20085927.
- Brown, E. J. (1985). "Interaction of Gram-Positive Microorganisms with Complement". Bacteria and Complement. Current Topics in Microbiology and Immunology. 121. Springer, Berlin, Heidelberg. pp. 159–187. doi:10.1007/978-3-642-45604-6_8. ISBN 9783642456060.
- Ram S, Lewis LA, Rice PA (2010). "Infections of people with complement deficiencies and patients who have undergone splenectomy". Clinical Microbiology Reviews. 23 (4): 740–80. doi:10.1128/CMR.00048-09. PMC 2952982. PMID 20930072.
- Lewis LA, Ram S (2014). "Meningococcal disease and the complement system". Virulence. 5 (1): 98–126. doi:10.4161/viru.26515. PMC 3916388. PMID 24104403.
- Dragon-Durey, Marie-Agnès; Frémeaux-Bacchi, Véronique (2005). "Atypical haemolytic uraemic syndrome and mutations in complement regulator genes". Springer Seminars in Immunopathology. 27 (3): 359–74. doi:10.1007/s00281-005-0003-2. PMID 16189652.
- Zipfel, Peter; Misselwitz, Joachim; Licht, Christoph; Skerka, Christine (2006). "The Role of Defective Complement Control in Hemolytic Uremic Syndrome". Seminars in Thrombosis and Hemostasis. 32 (2): 146–54. doi:10.1055/s-2006-939770. PMID 16575689.
- Mooijaart, Simon P.; Koeijvoets, Kristel M.C.; Sijbrands, Eric J.G.; Daha, Mohamed R.; Westendorp, Rudi G.J. (2007). "Complement Factor H polymorphism Y402H associates with inflammation, visual acuity, and cardiovascular mortality in the elderly population at large". Experimental Gerontology. 42 (11): 1116–22. doi:10.1016/j.exger.2007.08.001. PMID 17869048.
- Bradley, D T; Zipfel, P F; Hughes, A E (2011). "Complement in age-related macular degeneration: A focus on function". Eye. 25 (6): 683–93. doi:10.1038/eye.2011.37. PMC 3178140. PMID 21394116.
- Parker, C.; Omine, M.; Richards, S.; et al. (2005). "Diagnosis and Management of Paroxysmal Nocturnal Hemoglobinuria". Blood. 106 (12): 3699–3709. doi:10.1182/blood-2005-04-1717. PMC 1895106. PMID 16051736.
- "Complement Deficiencies Workup: Laboratory Studies, Imaging Studies, Other Tests". emedicine.medscape.com. Retrieved 2018-04-26.
- Datta, P.K.; Rappaport, J. (2006). "HIV and complement: Hijacking an immune defense". Biomedicine & Pharmacotherapy. 60 (9): 561–8. doi:10.1016/j.biopha.2006.07.087. PMID 16978830.
External links
