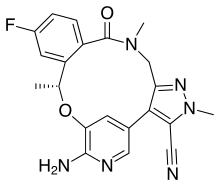
Lorlatinib
Lorlatinib, sold under the brand name Lorbrena in the United States, Canada, and Japan, and Lorviqua in the European Union, is an anti-cancer drug developed by Pfizer. It is an orally administered inhibitor of ALK and ROS1, two enzymes that play a role in the development of cancer.
 | |
| Clinical data | |
|---|---|
| Trade names | Lorbrena, Lorviqua |
| Other names | PF-6463922 |
| AHFS/Drugs.com | lorbrena |
| Routes of administration | By mouth |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 81% |
| Protein binding | 66% |
| Metabolism | Mainly CYP3A4 and UGT1A4 |
| Elimination half-life | 24 hrs (single dose) |
| Excretion | 48% urine (<1% unchanged), 41% faeces (9% unchanged) |
| Identifiers | |
IUPAC name
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.245.079 |
| Chemical and physical data | |
| Formula | C21H19FN6O2 |
| Molar mass | 406.421 g·mol−1 |
| 3D model (JSmol) | |
SMILES
| |
InChI
| |
Medical uses
Lortalinib is approved in the US and in Europe for the second- or third-line treatment of ALK-positive metastatic non-small-cell lung cancer (NSCLC).[1][2]
Contraindications
Lorlatinib must not be combined with strong inducers (i.e. activators) of the liver enzymes CYP3A4/5 if it can be avoided, as serious cases of liver toxicity have been observed under combination with the CYP3A4/5 inducer rifampicin.[3][4]
Side effects
The most common side effects in studies were high blood cholesterol (84% of patients), high blood triglycerides (67%), edema (55%), peripheral neuropathy (48%), cognitive effects (29%), fatigue (28%), weight gain (26%), and mood effects (23%). Serious side effects led to dose reduction in 23% of patients and in termination of lorlatinib treatment in 3% of patients.[3][4]
Interactions
Lorlatinib is metabolized by the enzymes CYP3A4/5. Therefore, CYP3A4/5 inducers such as rifampicin, carbamazepine or St John's wort decrease its concentrations in the blood plasma and can reduce its effectiveness. Additionally, the combination of lorlatinib with rifampicin showed liver toxicity in studies. Inhibitors of these enzymes such as ketoconazole or grapefruit juice increase lorlatinib plasma concentrations, leading to higher toxicity. Lorlatinib is also a (moderate) CYP3A4/5 inducer, so that drugs that are metabolized by these enzymes are broken down more quickly when combined with lorlatinib. Examples include midazolam and ciclosporin.[3][4]
Interactions via other enzymes have only been studied in vitro. According to these findings, lorlatinib may inhibit CYP2C9, UGT1A1 and several transport proteins, induce CYP2B6, and has probably no relevant effect on CYP1A2.[4]
Pharmacology
Mechanism of action
Lorlatinib is a small molecule kinase inhibitor of ALK and ROS1 as well as a number of other kinases. It is active in vitro against many mutated forms of ALK.[3]
Pharmacokinetics
The drug is swallowed in the form of tablets. It reaches highest blood plasma concentrations 1.2 hours after a single dose, or 2 hours after ingestion when taken regularly. Its absolute bioavailability is 80.8%. Intake with fatty food increases its availability by 5%, which is not considered clinically significant. When in the bloodstream, 66% of the substance are bound to plasma proteins.[3][4] Lorlatinib is able to cross the blood-brain barrier.[6]
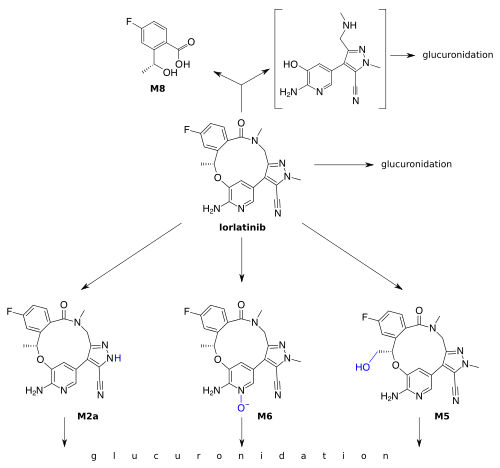
Lorlatinib is inactivated by oxidation, mainly through CYP3A4, and by glucuronidation, mainly through UGT1A4. Other CYPs and UGTs play a minor role. Lorlatinib and its metabolites are excreted with a half-life of 23.6 hours after a single dose; 47.7% into the urine (of which less than 1% in unchanged form), and 40.9% into the faeces (9.1% unchanged).[4]
Chemistry
Lorlatinib is a white to off-white powder. It has high solubility in 0.1 M hydrochloric acid and very low solubility at a pH over 4.5.[5]
History
Clinical studies
Several clinical trials are ongoing. A phase II trial comparing avelumab alone and in combination with lorlatinib or crizotinib for non-small-cell lung cancer is expected to be complete in late 2017. A phase II trial comparing lorlatinib with crizotinib is expected to be complete in mid-2018.[7] A phase II trial for treatment of ALK-positive or ROS1-positive non-small-cell lung cancer with central nervous system metastases is not expected to be complete until 2023.[8] Preclinical studies are investigating lorlatinib for treatment of neuroblastoma.
In 2017, Pfizer announced that lorlatinib was shown to have activity against lung and brain tumors in people with ALK or ROS1 positive advanced non-small-cell lung cancer.[9]
Approval
In 2015, FDA granted Pfizer orphan drug status for lorlatinib for the treatment of NSCLC.[10] In 2018, the FDA approved lortalinib for second- or third-line treatment of ALK-positive metastatic NSCLC.[1] In February 2019, the European CHMP of EMA recommended the granting of a conditional marketing authorisation.[11] In May 2019 the European Commission approved lorlatinib for the 28 countries of the EU, also as a second- or third-line treatment.[2]
References
- "FDA approves lorlatinib for second- or third-line treatment of ALK-positive metastatic NSCLC".
- "European Commission Approves LORVIQUA (lorlatinib) for Certain Adult Patients with Previously-Treated ALK-Positive Advanced Non-Small Cell Lung Cancer, PM Pfizer, May 7, 2019". pfizer.com. Retrieved 15 May 2019.
- FDA Professional Drug Information on Lorbrena.
- "Lorviqua: EPAR – Product Information" (PDF). European Medicines Agency. 2019-06-17.
- "Lorviqua: EPAR – Public assessment report" (PDF). European Medicines Agency. 2019-06-17.
- "NCI Drug Dictionary". National Cancer Institute.
- "A Study Of PF-06463922 An ALK/ROS1 Inhibitor In Patients With Advanced Non Small Cell Lung Cancer With Specific Molecular Alterations - Full Text View - ClinicalTrials.gov".
- "A Study of Lorlatinib in Advanced ALK and ROS1 Rearranged Lung Cancer With CNS Metastasis in the Absence of Measurable Extracranial Lesions - Full Text View - ClinicalTrials.gov".
- http://www.ascopost.com/News/58148
- http://drugspider.com/drug/lorlatinib
- "EMA Positive Opinion - Lorviqua, February 28, 2019". ema.europa.eu. Retrieved 15 May 2019.