Neuroblastoma
Neuroblastoma (NB) is a type of cancer that forms in certain types of nerve tissue.[1] It most frequently starts from one of the adrenal glands but can also develop in the neck, chest, abdomen, or spine.[1] Symptoms may include bone pain, a lump in the abdomen, neck, or chest, or a painless bluish lump under the skin.[1]
| Neuroblastoma | |
|---|---|
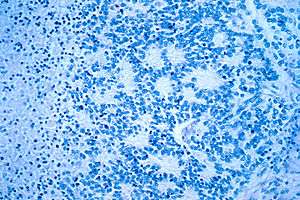 | |
| Microscopic view of a typical neuroblastoma with rosette formation | |
| Specialty | Oncology |
| Symptoms | Bone pain, lumps[1] |
| Usual onset | Under 5 years old[1] |
| Causes | Genetic mutation[1] |
| Diagnostic method | Tissue biopsy[1] |
| Treatment | Observation, surgery, radiation, chemotherapy, stem cell transplantation[1] |
| Prognosis | US five-year survival ~95% (< 1 year old), 68% (1–14 years old)[2] |
| Frequency | 1 in 7,000 children[2] |
| Deaths | 15% of deaths due to cancer in children[3] |
Typically, neuroblastoma occurs due to a genetic mutation occurring during early development.[4] Rarely, it may be due to a mutation inherited from a person's parents.[1] Environmental factors have not been found to be involved.[2] Diagnosis is based on a tissue biopsy.[1] Occasionally, it may be found in a baby by ultrasound during pregnancy.[1] At diagnosis, the cancer has usually already spread.[1] The cancer is divided into low-, intermediate-, and high-risk groups based on a child's age, cancer stage, and what the cancer looks like.[1]
Treatment and outcomes depends on the risk group a person is in.[1][4] Treatments may include observation, surgery, radiation, chemotherapy, or stem cell transplantation.[1] Low-risk disease in babies typically has a good outcome with surgery or simply observation.[4] In high-risk disease, chances of long-term survival, however, are less than 40%, despite aggressive treatment.[4]
Neuroblastoma is the most common cancer in babies and the third-most common cancer in children after leukemia and brain cancer.[4] About one in every 7,000 children is affected at some time.[2] About 90% of cases occur in children less than 5 years old, and it is rare in adults.[2][3] Of cancer deaths in children, about 15% are due to neuroblastoma.[3] The disease was first described in the 1800s.[5]
Signs and symptoms
The first symptoms of neuroblastoma are often vague, making diagnosis difficult. Fatigue, loss of appetite, fever, and joint pain are common. Symptoms depend on primary tumor locations and metastases if present:[6]
- In the abdomen, a tumor may cause a swollen belly and constipation.
- A tumor in the chest may cause breathing problems.
- A tumor pressing on the spinal cord may cause weakness, thus an inability to stand, crawl, or walk.
- Bone lesions in the legs and hips may cause pain and limping.
- A tumor in the bones around the eyes or orbits may cause distinct bruising and swelling.
- Infiltration of the bone marrow may cause pallor from anemia.
Neuroblastoma often spreads to other parts of the body before any symptoms are apparent, and 50 to 60% of all neuroblastoma cases present with metastases.[7]
The most common location for neuroblastoma to originate (i.e., the primary tumor) is in the adrenal glands. This occurs in 40% of localized tumors and in 60% of cases of widespread disease. Neuroblastoma can also develop anywhere along the sympathetic nervous system chain from the neck to the pelvis. Frequencies in different locations include: neck (1%), chest (19%), abdomen (30% nonadrenal), or pelvis (1%). In rare cases, no primary tumor can be discerned.[8]
Rare but characteristic presentations include transverse myelopathy (tumor spinal cord compression, 5% of cases), treatment-resistant diarrhea (tumor vasoactive intestinal peptide secretion, 4% of cases), Horner's syndrome (cervical tumor, 2.4% of cases), opsoclonus myoclonus syndrome[9] and ataxia (suspected paraneoplastic cause, 1.3% of cases), and hypertension (catecholamine secretion or kidney artery compression, 1.3% of cases).[10]
Cause
The cause of neuroblastoma is not well understood. The great majority of cases are sporadic and nonfamilial. About 1–2% of cases run in families and have been linked to specific gene mutations. Familial neuroblastoma in some cases is caused by rare germline mutations in the anaplastic lymphoma kinase (ALK) gene.[11] Germline mutations in the PHOX2B or KIF1B gene have been implicated in familial neuroblastoma, as well. Neuroblastoma is also a feature of neurofibromatosis type 1 and the Beckwith-Wiedemann syndrome.
MYCN oncogene amplification within the tumor is a common finding in neuroblastoma. The degree of amplification shows a bimodal distribution: either 3- to 10-fold, or 100- to 300-fold. The presence of this mutation is highly correlated to advanced stages of disease.[12]
Duplicated segments of the LMO1 gene within neuroblastoma tumor cells have been shown to increase the risk of developing an aggressive form of the cancer.[13]
Neuroblastoma has been linked to copy-number variation within the NBPF10 gene, which results in the 1q21.1 deletion syndrome or 1q21.1 duplication syndrome.[14]
Several risk factors have been proposed and are the subject of ongoing research. Due to characteristic early onset, many studies have focused on parental factors around conception and during gestation. Factors investigated have included occupation (i.e. exposure to chemicals in specific industries), smoking, alcohol consumption, use of medicinal drugs during pregnancy, and birth factors; however, results have been inconclusive.[15]
Other studies have examined possible links with atopy and exposure to infection early in life,[16] use of hormones and fertility drugs,[17] and maternal use of hair dye.[18][19]
Diagnosis
The diagnosis is usually confirmed by a surgical pathologist, taking into account the clinical presentation, microscopic findings, and other laboratory tests. It may arise from any neural crest element of the sympathetic nervous system (SNS).
Esthesioneuroblastoma, also known as olfactory neuroblastoma, is believed to arise from the olfactory epithelium and its classification remains controversial. However, since it is not a sympathetic nervous system malignancy, esthesioneuroblastoma is a distinct clinical entity and is not to be confused with neuroblastoma.[20][21]
Biochemistry
In about 90% of cases of neuroblastoma, elevated levels of catecholamines or their metabolites are found in the urine or blood. Catecholamines and their metabolites include dopamine, homovanillic acid (HVA), and/or vanillylmandelic acid (VMA).[22]
Imaging
Another way to detect neuroblastoma is the meta-iodobenzylguanidine scan, which is taken up by 90 to 95% of all neuroblastomas, often termed "mIBG-avid".[23] The mechanism is that mIBG is taken up by sympathetic neurons, and is a functioning analog of the neurotransmitter norepinephrine. When it is radio-iodinated with I-131 or I-123 (radioactive iodine isotopes), it is a very good radiopharmaceutical for diagnosis and monitoring of response to treatment for this disease. With a half-life of 13 hours, I-123 is the preferred isotope for imaging sensitivity and quality. I-131 has a half-life of 8 days and at higher doses is an effective therapy as targeted radiation against relapsed and refractory neuroblastoma.[24] As mIBG is not always taken up by neuroblastomas, researchers have explored in children with neuroblastoma whether another type of nuclear imaging, fluoro-deoxy-glucose - positron emission tomography, often termed "F-FDG-PET", might be useful.[25] Evidence suggests that this might be advisable to use in children with neuroblastoma for which mIBG does not work, but more research is needed in this area.[25]
Histology
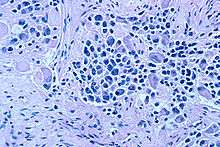
On microscopy, the tumor cells are typically described as small, round and blue, and rosette patterns (Homer Wright pseudorosettes) may be seen. Homer Wright pseudorosettes are tumor cells around the neuropil, not to be confused with a true rosettes, which are tumor cells around an empty lumen.[26] They are also distinct from the pseudorosettes of an ependymoma which consist of tumor cells with glial fibrillary acidic protein (GFAP)–positive processes tapering off toward a blood vessel (thus a combination of the two).[27] A variety of immunohistochemical stains are used by pathologists to distinguish neuroblastomas from histological mimics, such as rhabdomyosarcoma, Ewing's sarcoma, lymphoma and Wilms' tumor.[28]
Neuroblastoma is one of the peripheral neuroblastic tumors (pNTs) that have similar origins and show a wide pattern of differentiation ranging from benign ganglioneuroma to stroma-rich ganglioneuroblastoma with neuroblastic cells intermixed or in nodules, to highly malignant neuroblastoma. This distinction in the pre-treatment tumor pathology is an important prognostic factor, along with age and mitosis-karyorrhexis index (MKI). This pathology classification system (the Shimada system) describes "favorable" and "unfavorable" tumors by the International Neuroblastoma Pathology Committee (INPC) which was established in 1999 and revised in 2003.[29]
Staging
The "International Neuroblastoma Staging System" (INSS) established in 1986 and revised in 1988 stratifies neuroblastoma according to its anatomical presence at diagnosis:[30][31][32]
- Stage 1: Localized tumor confined to the area of origin.
- Stage 2A: Unilateral tumor with incomplete gross resection; identifiable ipsilateral and contralateral lymph node negative for tumor.
- Stage 2B: Unilateral tumor with complete or incomplete gross resection; with ipsilateral lymph node positive for tumor; identifiable contralateral lymph node negative for tumor.
- Stage 3: Tumor infiltrating across midline with or without regional lymph node involvement; or unilateral tumor with contralateral lymph node involvement; or midline tumor with bilateral lymph node involvement.
- Stage 4: Dissemination of tumor to distant lymph nodes, bone marrow, bone, liver, or other organs except as defined by Stage 4S.
- Stage 4S: Age <1 year old with localized primary tumor as defined in Stage 1 or 2, with dissemination limited to liver, skin, or bone marrow (less than 10 percent of nucleated bone marrow cells are tumors).
Although international agreement on staging (INSS) has been used, the need for an international consensus on risk assignment has also been recognized in order to compare similar cohorts in results of studies. Beginning in 2005, representatives of the major pediatric oncology cooperative groups have met to review data for 8,800 people with neuroblastoma treated in Europe, Japan, USA, Canada, and Australia between 1990 and 2002. This task force has proposed the International Neuroblastoma Risk Group (INRG) classification system. Retrospective studies revealed the high survival rate of 12–18 month old age group, previously categorized as high-risk, and prompted the decision to reclassify 12–18 month old children without N-myc (also commonly referred to as MYCN) amplification to intermediate risk category.[33]
The new INRG risk assignment will classify neuroblastoma at diagnosis based on a new International Neuroblastoma Risk Group Staging System (INRGSS):
- Stage L1: Localized disease without image-defined risk factors.
- Stage L2: Localized disease with image-defined risk factors.
- Stage M: Metastatic disease.
- Stage MS: Metastatic disease "special" where MS is equivalent to stage 4S.
The new risk stratification will be based on the new INRGSS staging system, age (dichotomized at 18 months), tumor grade, N-myc amplification, unbalanced 11q aberration, and ploidy into four pre-treatment risk groups: very low, low, intermediate, and high risk.[4][34]
Screening
Urine catecholamine level can be elevated in pre-clinical neuroblastoma. Screening asymptomatic infants at three weeks, six months, and one year has been performed in Japan, Canada, Austria and Germany since the 1980s.[35][36] Japan began screening six-month-olds for neuroblastoma via analysis of the levels of homovanillic acid and vanilmandelic acid in 1984. Screening was halted in 2004 after studies in Canada and Germany showed no reduction in deaths due to neuroblastoma, but rather caused an increase in diagnoses that would have disappeared without treatment, subjecting those infants to unnecessary surgery and chemotherapy.[37][38][39]
Treatment
When the lesion is localized, it is generally curable. However, long-term survival for children with advanced disease older than 18 months of age is poor despite aggressive multimodal therapy (intensive chemotherapy, surgery, radiation therapy, stem cell transplant, differentiation agent isotretinoin also called 13-cis-retinoic acid, and frequently immunotherapy[40] with anti-GD2 monoclonal antibody therapy).
Biologic and genetic characteristics have been identified, which, when added to classic clinical staging, has allowed assignment to risk groups for planning treatment intensity.[41] These criteria include the age of the person, extent of disease spread, microscopic appearance, and genetic features including DNA ploidy and N-myc oncogene amplification (N-myc regulates microRNAs[42]), into low, intermediate, and high risk disease. A recent biology study (COG ANBL00B1) analyzed 2687 people with neuroblastoma and the spectrum of risk assignment was determined: 37% of neuroblastoma cases are low risk, 18% are intermediate risk, and 45% are high risk.[43] (There is some evidence that the high- and low-risk types are caused by different mechanisms, and are not merely two different degrees of expression of the same mechanism.)[44]
The therapies for these different risk categories are very different.
- Low-risk disease can frequently be observed without any treatment at all or cured with surgery alone.[45]
- Intermediate-risk disease is treated with surgery and chemotherapy.[46]
- High-risk neuroblastoma is treated with intensive chemotherapy, surgery, radiation therapy, bone marrow / hematopoietic stem cell transplantation,[47] biological-based therapy with 13-cis-retinoic acid (isotretinoin or Accutane)[48] and antibody therapy usually administered with the cytokines GM-CSF and IL-2.[49] A meta analysis has found evidence that in children with high-risk neuroblastoma, treatment with myeloablative therapy improves event-free survival but may increase the risk of side effects such as kidney problems when compared to conventional chemotherapy.[50]
With current treatments, people with low and intermediate risk disease have an excellent prognosis with cure rates above 90% for low risk and 70–90% for intermediate risk. In contrast, therapy for high-risk neuroblastoma the past two decades resulted in cures only about 30% of the time.[51] The addition of antibody therapy has raised survival rates for high-risk disease significantly. In March 2009 an early analysis of a Children's Oncology Group (COG) study with 226 people that are high-risk showed that two years after stem cell transplant 66% of the group randomized to receive ch14.18 antibody with GM-CSF and IL-2 were alive and disease-free compared to only 46% in the group that did not receive the antibody. The randomization was stopped so all people enrolling on the trial will receive the antibody therapy.[52]
Chemotherapy agents used in combination have been found to be effective against neuroblastoma. Agents commonly used in induction and for stem cell transplant conditioning are platinum compounds (cisplatin, carboplatin), alkylating agents (cyclophosphamide, ifosfamide, melphalan), topoisomerase II inhibitor (etoposide), anthracycline antibiotics (doxorubicin) and vinca alkaloids (vincristine). Some newer regimens include topoisomerase I inhibitors (topotecan and irinotecan) in induction which have been found to be effective against recurrent disease.
Prognosis
Between 20% and 50% of high-risk cases do not respond adequately to induction high-dose chemotherapy and are progressive or refractory.[53][54] Relapse after completion of frontline therapy is also common. Further treatment is available in phase I and phase II clinical trials that test new agents and combinations of agents against neuroblastoma, but the outcome remains very poor for relapsed high-risk disease.[55]
Most long-term survivors alive today had low or intermediate risk disease and milder courses of treatment compared to high-risk disease. The majority of survivors have long-term effects from the treatment. Survivors of intermediate and high-risk treatment often experience hearing loss, growth reduction, thyroid function disorders, learning difficulties, and greater risk of secondary cancers affect survivors of high-risk disease.[56][57] An estimated two of three survivors of childhood cancer will ultimately develop at least one chronic and sometimes life-threatening health problem within 20 to 30 years after the cancer diagnosis.[58][59][60]
Cytogenetic profiles
Based on a series of 493 neuroblastoma samples, it has been reported that overall genomic pattern, as tested by array-based karyotyping, is a predictor of outcome in neuroblastoma:[61]
- Tumors presenting exclusively with whole chromosome copy number changes were associated with excellent survival.
- Tumors presenting with any kind of segmental chromosome copy number changes were associated with a high risk of relapse.
- Within tumors showing segmental alterations, additional independent predictors of decreased overall survival were N-myc amplification, 1p and 11q deletions, and 1q gain.
Earlier publications categorized neuroblastomas into three major subtypes based on cytogenetic profiles:[62][63]
- Subtype 1: favorable neuroblastoma with near triploidy and a predominance of numerical gains and losses, mostly representing non-metastatic NB stages 1, 2 and 4S.
- Subtypes 2A and 2B: found in unfavorable widespread neuroblastoma, stages 3 and 4, with 11q loss and 17q gain without N-myc amplification (subtype 2A) or with N-myc amplification often together with 1p deletions and 17q gain (subtype 2B).
Virtual karyotyping can be performed on fresh or paraffin-embedded tumors to assess copy number at these loci. SNP array virtual karyotyping is preferred for tumor samples, including neuroblastomas, because they can detect copy neutral loss of heterozygosity (acquired uniparental disomy). Copy neutral LOH can be biologically equivalent to a deletion and has been detected at key loci in neuroblastoma.[64] ArrayCGH, FISH, or conventional cytogenetics cannot detect copy neutral LOH.
Epidemiology
Neuroblastoma comprises 6–10% of all childhood cancers, and 15% of cancer deaths in children. The annual mortality rate is 10 per million children in the 0- to 4-year-old age group, and 4 per million in the 4- to 9-year old age group.[65]
The highest number of cases is in the first year of life, and some cases are congenital. The age range is broad, including older children and adults,[66] but only 10% of cases occur in people older than 5 years of age.[23] A large European study reported less than 2% of over 4000 neuroblastoma cases were over 18 years old.[67]
History

In 1864 German physician Rudolf Virchow was the first to describe an abdominal tumor in a child as a "glioma". The characteristics of tumors from the sympathetic nervous system and the adrenal medulla were then noted in 1891 by German pathologist Felix Marchand.[68][69] In 1901 the distinctive presentation of stage 4S in infants (liver but no bone metastases) was described by William Pepper. In 1910 James Homer Wright understood the tumor to originate from primitive neural cells, and named it neuroblastoma. He also noted the circular clumps of cells in bone marrow samples which are now termed "Homer Wright rosettes". Of note, "Homer-Wright" with a hyphen is grammatically incorrect, as the eponym refers to just Dr. Wright.[70]
Society and culture
Legislative efforts
U.S. Representative Chet Edwards of Waco, Texas, successfully introduced legislation to earmark $150 million toward a cure for neuroblastoma and other cancers. The measure was signed into law in July 2008 by U.S. President George W. Bush. Edwards was inspired in the endeavor by the illness and subsequent death of Erin Channing Buenger (1997–2009) of Bryan, daughter of one of his constituents, Walter L. Buenger, head of the history department at Texas A&M University.[71]
Research
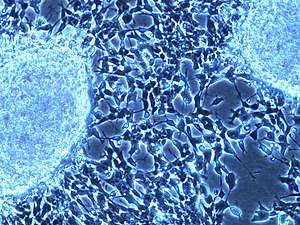
Preclinical models
Neuroblastoma patient derived tumor xenografts (PDXs) have been created by orthotopic implantation of tumor samples into immunodeficient mice.[72] PDX models have several advantages over conventional cancer cell lines (CCL)s.[73] Neuroblastoma PDXs retain the genetic hallmarks of their corresponding tumors and PDXs display infiltrative growth and metastasis to distant organs.[72] PDX models are more predictive of clinical outcome as compared to conventional cancer cell line xenografts.[74] Neuroblastoma PDXs might thus serve as clinically relevant models to identify effective compounds against neuroblastoma.[72]
Treatments
Recent focus has been to reduce therapy for low and intermediate risk neuroblastoma while maintaining survival rates at 90%.[75] A study of 467 people that are at intermediate risk enrolled in A3961 from 1997 to 2005 confirmed the hypothesis that therapy could be successfully reduced for this risk group. Those with favorable characteristics (tumor grade and response) received four cycles of chemotherapy, and those with unfavorable characteristics received eight cycles, with three-year event free survival and overall survival stable at 90% for the entire cohort. Future plans are to intensify treatment for those people with aberration of 1p36 or 11q23 chromosomes as well as for those who lack early response to treatment.[76][77]
By contrast, focus the past 20 years or more has been to intensify treatment for high-risk neuroblastoma. Chemotherapy induction variations, timing of surgery, stem cell transplant regimens, various delivery schemes for radiation, and use of monoclonal antibodies and retinoids to treat minimal residual disease continue to be examined. Recent phase III clinical trials with randomization have been carried out to answer these questions to improve survival of high-risk disease:
Refractory and relapsed neuroblastoma
Chemotherapy with topotecan and cyclophosphamide is frequently used in refractory setting and after relapse.[78]
A haploidentical stem cell transplant, that is, donor cells derived from parents, is being studied in those with refractory or relapsing neuroblastoma as stem cells from the person themselves is not useful.[79]
References
- "Neuroblastoma Treatment". National Cancer Institute. 20 January 2016. Archived from the original on 10 November 2016. Retrieved 9 November 2016.
- "Neuroblastoma Treatment". National Cancer Institute. 25 August 2016. Archived from the original on 10 November 2016. Retrieved 10 November 2016.
- World Cancer Report 2014. World Health Organization. 2014. Chapter 5.16. ISBN 978-9283204299. Archived from the original on 2016-09-19.
- Maris JM, Hogarty MD, Bagatell R, Cohn SL (June 2007). "Neuroblastoma". Lancet. 369 (9579): 2106–20. doi:10.1016/S0140-6736(07)60983-0. PMID 17586306.
- Olson, James Stuart (1989). The History of Cancer: An Annotated Bibliography. ABC-CLIO. p. 177. ISBN 9780313258893. Archived from the original on 2017-09-10.
- Wheeler, Kate (January 1, 2013). "Neuroblastoma in children". Macmillan. Archived from the original on October 5, 2015.
- "Neuroblastoma: Pediatric Cancers: Merck Manual Professional". Archived from the original on 2007-12-18. Retrieved 2008-01-01.
- Friedman GK, Castleberry RP (December 2007). "Changing trends of research and treatment in infant neuroblastoma". Pediatric Blood & Cancer. 49 (7 Suppl): 1060–5. doi:10.1002/pbc.21354. PMID 17943963.
- Rothenberg AB, Berdon WE, D'Angio GJ, Yamashiro DJ, Cowles RA (July 2009). "The association between neuroblastoma and opsoclonus-myoclonus syndrome: a historical review". Pediatric Radiology. 39 (7): 723–6. doi:10.1007/s00247-009-1282-x. PMID 19430769.
- Cheung, Nai-Kong (2005). Neuroblastoma. Springer-Verlag. pp. 66–7. ISBN 978-3-540-40841-3.
- Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. (October 2008). "Identification of ALK as a major familial neuroblastoma predisposition gene". Nature. 455 (7215): 930–5. Bibcode:2008Natur.455..930M. doi:10.1038/nature07261. PMC 2672043. PMID 18724359.
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM (June 1984). "Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage". Science. 224 (4653): 1121–4. Bibcode:1984Sci...224.1121B. doi:10.1126/science.6719137. PMID 6719137.
- Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, et al. (January 2011). "Integrative genomics identifies LMO1 as a neuroblastoma oncogene". Nature. 469 (7329): 216–20. Bibcode:2011Natur.469..216W. doi:10.1038/nature09609. PMC 3320515. PMID 21124317. Lay summary – Children's Hospital of Philadelphia (November 30, 2010).
- Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, et al. (June 2009). "Copy number variation at 1q21.1 associated with neuroblastoma". Nature. 459 (7249): 987–91. Bibcode:2009Natur.459..987D. doi:10.1038/nature08035. PMC 2755253. PMID 19536264.
- Olshan AF, Bunin GR (2000). "Epidemiology of Neuroblastoma". In Brodeur GM, Sawada T, Tsuchida Y, et al. (eds.). Neuroblastoma. Amsterdam: Elsevier. pp. 33–9. ISBN 978-0-444-50222-3.
- Menegaux F, Olshan AF, Neglia JP, Pollock BH, Bondy ML (May 2004). "Day care, childhood infections, and risk of neuroblastoma". American Journal of Epidemiology. 159 (9): 843–51. doi:10.1093/aje/kwh111. PMC 2080646. PMID 15105177.
- Olshan AF, Smith J, Cook MN, Grufferman S, Pollock BH, Stram DO, et al. (November 1999). "Hormone and fertility drug use and the risk of neuroblastoma: a report from the Children's Cancer Group and the Pediatric Oncology Group". American Journal of Epidemiology. 150 (9): 930–8. doi:10.1093/oxfordjournals.aje.a010101. PMID 10547138.
- McCall EE, Olshan AF, Daniels JL (August 2005). "Maternal hair dye use and risk of neuroblastoma in offspring". Cancer Causes & Control. 16 (6): 743–8. doi:10.1007/s10552-005-1229-y. PMID 16049813.
- Heck JE, Ritz B, Hung RJ, Hashibe M, Boffetta P (March 2009). "The epidemiology of neuroblastoma: a review". Paediatric and Perinatal Epidemiology. 23 (2): 125–43. doi:10.1111/j.1365-3016.2008.00983.x. PMID 19159399.
- Esthesioneuroblastoma at eMedicine
- Cheung, Nai-Kong (2005). Neuroblastoma. Springer-Verlag. p. 73. ISBN 978-3-540-40841-3.
- Strenger V, Kerbl R, Dornbusch HJ, Ladenstein R, Ambros PF, Ambros IM, Urban C (May 2007). "Diagnostic and prognostic impact of urinary catecholamines in neuroblastoma patients". Pediatric Blood & Cancer. 48 (5): 504–9. doi:10.1002/pbc.20888. PMID 16732582.
- Howman-Giles R, Shaw PJ, Uren RF, Chung DK (July 2007). "Neuroblastoma and other neuroendocrine tumors". Seminars in Nuclear Medicine. 37 (4): 286–302. doi:10.1053/j.semnuclmed.2007.02.009. PMID 17544628.
- Pashankar FD, O'Dorisio MS, Menda Y (January 2005). "MIBG and somatostatin receptor analogs in children: current concepts on diagnostic and therapeutic use". Journal of Nuclear Medicine. 46 Suppl 1 (Suppl 1): 55S–61S. PMID 15653652.
- Bleeker G, Tytgat GA, Adam JA, Caron HN, Kremer LC, Hooft L, van Dalen EC (September 2015). "123I-MIBG scintigraphy and 18F-FDG-PET imaging for diagnosing neuroblastoma". The Cochrane Database of Systematic Reviews (9): CD009263. doi:10.1002/14651858.cd009263.pub2. PMC 4621955. PMID 26417712.
- Robbins and Cotran pathologic basis of disease (9 ed.). Elsevier. 2015. ISBN 978-1455726134.
- Ependymoma at eMedicine
- Carter RL, al-Sams SZ, Corbett RP, Clinton S (May 1990). "A comparative study of immunohistochemical staining for neuron-specific enolase, protein gene product 9.5 and S-100 protein in neuroblastoma, Ewing's sarcoma and other round cell tumours in children". Histopathology. 16 (5): 461–7. doi:10.1111/j.1365-2559.1990.tb01545.x. PMID 2163356.
- Peuchmaur M, d'Amore ES, Joshi VV, Hata J, Roald B, Dehner LP, et al. (November 2003). "Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular". Cancer. 98 (10): 2274–81. doi:10.1002/cncr.11773. PMID 14601099.
- "Neuroblastoma Treatment—National Cancer Institute". 1980-01-01. Archived from the original on 2008-10-02. Retrieved 2008-07-30.
- Brodeur GM, Seeger RC, Barrett A, Berthold F, Castleberry RP, D'Angio G, et al. (December 1988). "International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma" (PDF). Journal of Clinical Oncology. 6 (12): 1874–81. doi:10.1200/JCO.1988.6.12.1874. PMID 3199170.
- Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. (August 1993). "Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment". Journal of Clinical Oncology. 11 (8): 1466–77. doi:10.1200/JCO.1993.11.8.1466. PMID 8336186.
- Schmidt ML, Lal A, Seeger RC, Maris JM, Shimada H, O'Leary M, et al. (September 2005). "Favorable prognosis for patients 12 to 18 months of age with stage 4 nonamplified MYCN neuroblastoma: a Children's Cancer Group Study". Journal of Clinical Oncology. 23 (27): 6474–80. doi:10.1200/JCO.2005.05.183. PMID 16116154.
- Cohn SL, London WB, Monclair T, Matthay KK, Ambros PF, Pearson AD (2007). "Update on the development of the international neuroblastoma risk group (INRG) classification schema". Journal of Clinical Oncology. 25 (18 Suppl): 9503. doi:10.1200/jco.2007.25.18_suppl.9503 (inactive 2019-11-18). Archived from the original on 2016-01-10.
- Woods WG, Gao RN, Shuster JJ, Robison LL, Bernstein M, Weitzman S, et al. (April 2002). "Screening of infants and mortality due to neuroblastoma". The New England Journal of Medicine. 346 (14): 1041–6. doi:10.1056/NEJMoa012387. PMID 11932470.
- Schilling FH, Spix C, Berthold F, Erttmann R, Sander J, Treuner J, Michaelis J (July 2003). "Children may not benefit from neuroblastoma screening at 1 year of age. Updated results of the population based controlled trial in Germany". Cancer Letters. 197 (1–2): 19–28. doi:10.1016/S0304-3835(03)00077-6. PMID 12880955.
- Tsubono Y, Hisamichi S (May 2004). "A halt to neuroblastoma screening in Japan". The New England Journal of Medicine. 350 (19): 2010–1. doi:10.1056/NEJM200405063501922. PMID 15128908.
- "Neuroblastoma Screening". National Cancer Institute. 1980-01-01. Archived from the original on 2008-10-01. Retrieved 2008-07-30.
- Darshak Sanghavi, "Screen Alert: How an Ounce of RX Prevention can Cause a Pound of Hurt" Archived 2006-12-01 at the Wayback Machine, Slate magazine, November 28, 2006
- Johnson E, Dean SM, Sondel PM (December 2007). "Antibody-based immunotherapy in high-risk neuroblastoma". Expert Reviews in Molecular Medicine. 9 (34): 1–21. doi:10.1017/S1462399407000518. PMID 18081947.
- Brodeur GM (March 2003). "Neuroblastoma: biological insights into a clinical enigma". Nature Reviews. Cancer. 3 (3): 203–16. doi:10.1038/nrc1014. PMID 12612655.
- Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, et al. (February 2008). "MYCN regulates oncogenic MicroRNAs in neuroblastoma". International Journal of Cancer. 122 (3): 699–704. doi:10.1002/ijc.23153. PMID 17943719.
- "Translating Neuroblastoma Genomics to the Clinic—J. Maris presentation ASCO 2007". Archived from the original on 2009-01-02. Retrieved 2008-01-13.
- Gisselsson D, Lundberg G, Ora I, Höglund M (September 2007). "Distinct evolutionary mechanisms for genomic imbalances in high-risk and low-risk neuroblastomas". Journal of Carcinogenesis. 6: 15. doi:10.1186/1477-3163-6-15. PMC 2042979. PMID 17897457.
- "Neuroblastoma Treatment". National Cancer Institute. 1980-01-01. Archived from the original on 2008-05-03. Retrieved 2008-02-02.
- Haase GM, Perez C, Atkinson JB (March 1999). "Current aspects of biology, risk assessment, and treatment of neuroblastoma". Seminars in Surgical Oncology. 16 (2): 91–104. doi:10.1002/(SICI)1098-2388(199903)16:2<91::AID-SSU3>3.0.CO;2-1. PMID 9988866.
- Fish JD, Grupp SA (January 2008). "Stem cell transplantation for neuroblastoma". Bone Marrow Transplantation. 41 (2): 159–65. doi:10.1038/sj.bmt.1705929. PMC 2892221. PMID 18037943.
- Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, et al. (October 1999). "Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group". The New England Journal of Medicine. 341 (16): 1165–73. doi:10.1056/NEJM199910143411601. PMID 10519894.
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. (September 2010). "Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma". The New England Journal of Medicine. 363 (14): 1324–34. doi:10.1056/NEJMoa0911123. PMC 3086629. PMID 20879881.
- Yalçin B, Kremer LC, van Dalen EC (October 2015). "High-dose chemotherapy and autologous haematopoietic stem cell rescue for children with high-risk neuroblastoma". The Cochrane Database of Systematic Reviews (10): CD006301. doi:10.1002/14651858.cd006301.pub4. PMID 26436598.
- "Neuroblastoma Treatment". National Cancer Institute. 1980-01-01. Archived from the original on 2008-10-02. Retrieved 2008-07-30.
- Yu AL, Gilman MF, Ozkaynak WB, London S, Kreissman HX, Chen KK, Matthay SL, Cohn JM, Maris JM, Sondel PM (2009). "A phase III randomized trial of the chimeric anti-GD2 antibody ch14.18 with GM-CSF and IL2 as immunotherapy following dose intensive chemotherapy for high-risk neuroblastoma: Childrens Oncology Group (COG) study ANBL0032". Journal of Clinical Oncology. 27 (15 Suppl): 10067z. Archived from the original on 2016-01-10. Retrieved 2015-09-10.
- Kushner BH, Kramer K, LaQuaglia MP, Modak S, Yataghene K, Cheung NK (December 2004). "Reduction from seven to five cycles of intensive induction chemotherapy in children with high-risk neuroblastoma". Journal of Clinical Oncology. 22 (24): 4888–92. doi:10.1200/JCO.2004.02.101. PMID 15611504.
- Kreissman SG, Villablanca JG, Diller L, London WB, Maris JM, Park JR, Reynolds CP, von Allmen D, Cohn SL, Matthay KK (2007). "Response and toxicity to a dose-intensive multi-agent chemotherapy induction regimen for high risk neuroblastoma (HR-NB): A Children's Oncology Group (COG A3973) study". Journal of Clinical Oncology. 25 (18 Suppl): 9505. doi:10.1200/jco.2007.25.18_suppl.9505 (inactive 2019-11-18). Archived from the original on 2016-01-10.
- Ceschel S, Casotto V, Valsecchi MG, Tamaro P, Jankovic M, Hanau G, et al. (October 2006). "Survival after relapse in children with solid tumors: a follow-up study from the Italian off-therapy registry". Pediatric Blood & Cancer. 47 (5): 560–6. doi:10.1002/pbc.20726. PMID 16395684.
- Gurney JG, Tersak JM, Ness KK, Landier W, Matthay KK, Schmidt ML (November 2007). "Hearing loss, quality of life, and academic problems in long-term neuroblastoma survivors: a report from the Children's Oncology Group". Pediatrics. 120 (5): e1229–36. doi:10.1542/peds.2007-0178. PMID 17974716.
- Trahair TN, Vowels MR, Johnston K, Cohn RJ, Russell SJ, Neville KA, et al. (October 2007). "Long-term outcomes in children with high-risk neuroblastoma treated with autologous stem cell transplantation". Bone Marrow Transplantation. 40 (8): 741–6. doi:10.1038/sj.bmt.1705809. PMID 17724446.
- Mozes, Alan (February 21, 2007). "Childhood Cancer Survivors Face Increased Sarcoma Risk". HealthDay. Archived from the original on September 8, 2015.
- Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. (October 2006). "Chronic health conditions in adult survivors of childhood cancer". The New England Journal of Medicine. 355 (15): 1572–82. doi:10.1056/NEJMsa060185. PMID 17035650.
- Laverdière C, Liu Q, Yasui Y, Nathan PC, Gurney JG, Stovall M, et al. (August 2009). "Long-term outcomes in survivors of neuroblastoma: a report from the Childhood Cancer Survivor Study". Journal of the National Cancer Institute. 101 (16): 1131–40. doi:10.1093/jnci/djp230. PMC 2728747. PMID 19648511.
- Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, et al. (March 2009). "Overall genomic pattern is a predictor of outcome in neuroblastoma" (PDF). Journal of Clinical Oncology. 27 (7): 1026–33. doi:10.1200/JCO.2008.16.0630. PMID 19171713.
- Vandesompele J, Baudis M, De Preter K, Van Roy N, Ambros P, Bown N, et al. (April 2005). "Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma" (PDF). Journal of Clinical Oncology. 23 (10): 2280–99. doi:10.1200/JCO.2005.06.104. PMID 15800319.
- Michels E, Vandesompele J, Hoebeeck J, Menten B, De Preter K, Laureys G, et al. (2006). "Genome wide measurement of DNA copy number changes in neuroblastoma: dissecting amplicons and mapping losses, gains and breakpoints". Cytogenetic and Genome Research. 115 (3–4): 273–82. doi:10.1159/000095924. PMID 17124410.
- Carén H, Erichsen J, Olsson L, Enerbäck C, Sjöberg RM, Abrahamsson J, et al. (July 2008). "High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: four cases of homozygous deletions of the CDKN2A gene". BMC Genomics. 9: 353. doi:10.1186/1471-2164-9-353. PMC 2527340. PMID 18664255.
- Brodeur GM, Hogarty MD, Mosse YP, Maris JM (1997). "Neuroblastoma". In Pizzo PA, Poplack DG (eds.). Principles and Practice of Pediatric Oncology (6th ed.). pp. 886–922. ISBN 978-1-60547-682-7.
- Franks LM, Bollen A, Seeger RC, Stram DO, Matthay KK (May 1997). "Neuroblastoma in adults and adolescents: an indolent course with poor survival". Cancer. 79 (10): 2028–35. doi:10.1002/(SICI)1097-0142(19970515)79:10<2028::AID-CNCR26>3.0.CO;2-V. PMID 9149032.
- Ladenstein R, Pötschger U, Hartman O, Pearson AD, Klingebiel T, Castel V, et al. (June 2008). "28 years of high-dose therapy and SCT for neuroblastoma in Europe: lessons from more than 4000 procedures". Bone Marrow Transplantation. 41 Suppl 2 (Suppl 2): S118–27. doi:10.1038/bmt.2008.69. PMID 18545256.
- Berthold F, Simon T (2006). "Clinical Presentation". In Cheung NV, Cohn SL (eds.). Neuroblastoma. Springer. pp. 63–85. ISBN 978-3-540-26616-7.
- Beckwith JB, Perrin EV (December 1963). "In Situ Neuroblastomas: A Contribution to the Natural History of Neural Crest Tumors". The American Journal of Pathology. 43: 1089–104. PMC 1949785. PMID 14099453.
- Rothenberg AB, Berdon WE, D'Angio GJ, Yamashiro DJ, Cowles RA (February 2009). "Neuroblastoma-remembering the three physicians who described it a century ago: James Homer Wright, William Pepper, and Robert Hutchison". Pediatric Radiology. 39 (2): 155–60. doi:10.1007/s00247-008-1062-z. PMID 19034443.
- "Erin Buenger had a zest for living life fully". The Bryan College Station Eagle. April 12, 2009. Archived from the original on June 11, 2011.
- Braekeveldt N, Wigerup C, Gisselsson D, Mohlin S, Merselius M, Beckman S, et al. (March 2015). "Neuroblastoma patient-derived orthotopic xenografts retain metastatic patterns and geno- and phenotypes of patient tumours". International Journal of Cancer. 136 (5): E252–61. doi:10.1002/ijc.29217. PMC 4299502. PMID 25220031.
- Malaney P, Nicosia SV, Davé V (March 2014). "One mouse, one patient paradigm: New avatars of personalized cancer therapy". Cancer Letters. 344 (1): 1–12. doi:10.1016/j.canlet.2013.10.010. PMC 4092874. PMID 24157811.
- Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. (April 2012). "Patient-derived tumour xenografts as models for oncology drug development". Nature Reviews. Clinical Oncology. 9 (6): 338–50. doi:10.1038/nrclinonc.2012.61. PMC 3928688. PMID 22508028.
- "Neuroblastoma Committee—Current Focus of Research". Archived from the original on September 25, 2006. Retrieved 2008-01-13.
- Baker DL, Schmidt ML, Cohn SL, Maris JM, London WB, Buxton A, et al. (September 2010). "Outcome after reduced chemotherapy for intermediate-risk neuroblastoma". The New England Journal of Medicine. 363 (14): 1313–23. doi:10.1200/jco.2007.25.18_suppl.9504 (inactive 2019-11-18). PMID 20879880. Archived from the original on 2013-01-13.
- Baker DL, Schmidt ML, Cohn SL, Maris JM, London WB, Buxton A, et al. (September 2010). "Outcome after reduced chemotherapy for intermediate-risk neuroblastoma". The New England Journal of Medicine. 363 (14): 1313–23. doi:10.1056/NEJMoa1001527. PMC 2993160. PMID 20879880.
- Morgenstern DA, Baruchel S, Irwin MS (July 2013). "Current and future strategies for relapsed neuroblastoma: challenges on the road to precision therapy". Journal of Pediatric Hematology/Oncology. 35 (5): 337–47. doi:10.1097/MPH.0b013e318299d637. PMID 23703550.
- Illhardt T, Toporski J, Feuchtinger T, Turkiewicz D, Teltschik HM, Ebinger M, et al. (May 2018). "Haploidentical Stem Cell Transplantation for Refractory/Relapsed Neuroblastoma". Biology of Blood and Marrow Transplantation. Elsevier BV. 24 (5): 1005–1012. doi:10.1016/j.bbmt.2017.12.805. PMID 29307718.