Glomerulonephritis
Glomerulonephritis (GN) is a term used to refer to several kidney diseases (usually affecting both kidneys). Many of the diseases are characterised by inflammation either of the glomeruli or of the small blood vessels in the kidneys, hence the name,[1] but not all diseases necessarily have an inflammatory component.
| Glomerulonephritis | |
|---|---|
| Other names | Glomerular nephritis |
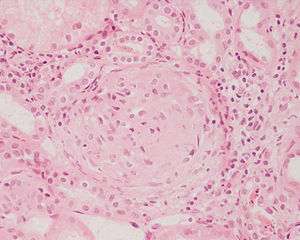 | |
| Photomicrograph of a kidney biopsy from a person with crescentic glomerulonephritis showing prominent fibrocellular crescent formation and moderate mesangial proliferation in a glomerulus. Hematoxylin and eosin stain. | |
| Specialty | Nephrology |
As it is not strictly a single disease, its presentation depends on the specific disease entity: it may present with isolated hematuria and/or proteinuria (blood or protein in the urine); or as a nephrotic syndrome, a nephritic syndrome, acute kidney injury, or chronic kidney disease.
They are categorized into several different pathological patterns, which are broadly grouped into non-proliferative or proliferative types. Diagnosing the pattern of GN is important because the outcome and treatment differs in different types. Primary causes are intrinsic to the kidney. Secondary causes are associated with certain infections (bacterial, viral or parasitic pathogens), drugs, systemic disorders (SLE, vasculitis), or diabetes.
Signs and symptoms
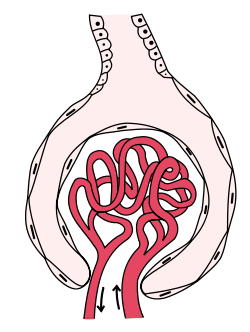
Glomerulonephritis refers to an inflammation of the glomerulus, which is the unit involved in filtration in the kidney. This inflammation typically results in one or both of the nephrotic or nephritic syndromes.[2]:500
Nephrotic syndrome
The nephrotic syndrome is characterised by the finding of edema in a person with increased protein in the urine and decreased protein in the blood, with increased fat in the blood. Inflammation that affects the cells surrounding the glomerulus, podocytes, increases the permeability to proteins, resulting in an increase in excreted proteins. When the amount of proteins excreted in the urine exceeds the liver's ability to compensate, fewer proteins are detected in the blood – in particular albumin, which makes up the majority of circulating proteins. With decreased proteins in the blood, there is a decrease in the oncotic pressure of the blood. This results in edema, as the oncotic pressure in tissue remains the same. Although decreased intravascular oncotic (i.e. osmotic) pressure partially explains the patient's edema, more recent studies have shown that extensive sodium retention in the distal nephron (collecting duct) is the predominant cause of water retention and edema in the nephrotic syndrome.[3] This is worsened by the secretion of the hormone aldosterone by the adrenal gland, which is secreted in response to the decrease in circulating blood and causes sodium and water retention. Hyperlipidemia is thought to be a result of the increased activity of the liver.[4]:549
Nephritic syndrome
The nephritic syndrome is characterised by blood in the urine (especially Red blood cell casts with dysmorphic red blood cells) and a decrease in the amount of urine in the presence of hypertension. In this syndrome, inflammatory damage to cells lining the glomerulus are thought to result in destruction of the epithelial barrier, leading to blood being found in the urine. At the same time, reactive changes, e.g. proliferation of mesangial cells, may result in a decrease in kidney blood flow, resulting in a decrease in the production of urine. The renin–angiotensin system may be subsequently activated, because of the decrease in perfusion of juxtaglomerular apparatus, which may result in hypertension.[4]:554
Nonproliferative
This is characterised by forms of glomerulonephritis in which the number of cells is not changed. These forms usually result in the nephrotic syndrome. Causes include:
Minimal change disease
Minimal change disease is characterised as a cause of nephrotic syndrome without visible changes in the glomerulus on microscopy. Minimal change disease typically presents with edema, an increase in proteins passed from urine and decrease in blood protein levels, and an increase in circulating lipids (i.e., nephrotic syndrome) and is the most common cause of the nephrotic syndrome in children. Although no changes may be visible by light microscopy, changes on electron microscopy within the glomerules may show a fusion of the foot processes of the podocytes (cells lining the basement membrane of the capillaries of glomerulus). It is typically managed with corticosteroids and does not progress to chronic kidney disease.[2]:500[4]:550
Focal segmental glomerulosclerosis
Focal segmental glomerulosclerosis is characterised by a sclerosis of segments of some glomerules. It is likely to present as a nephrotic syndrome. This form of glomerulonephritis may be associated with conditions such as HIV and heroin abuse, or inherited as Alport syndrome. The cause of about 20–30% of focal-segmental glomerulosclerosis is unknown. On microscopy, affected glomerules may show an increase in hyalin, a pink and homogenous material, fat cells, an increase in the mesangial matrix and collagen. Treatment may involve corticosteroids, but up to half of people with focal segmental glomerulonephritis continue to have progressive deterioration of kidney function, ending in kidney failure.[4]:550–551
Membranous glomerulonephritis
Membranous glomerulonephritis may cause either nephrotic or a nephritic picture. About two-thirds are associated with auto-antibodies to phospholipase A2 receptor, but other associations include cancers of the lung and bowel, infections such as hepatitis B and malaria, drugs including penicillamine, and connective tissue diseases such as systemic lupus erythematosus. Individuals with cerebral shunts are at risk of developing shunt nephritis, which frequently produces MGN.
Microscopically, MGN is characterized by a thickened glomerular basement membrane without a hyperproliferation of the glomerular cells. Immunofluorescence demonstrates diffuse granular uptake of IgG. The basement membrane may completely surround the granular deposits, forming a "spike and dome" pattern. Tubules also display the symptoms of a typical Type III hypersensitivity reaction, which causes the endothelial cells to proliferate, which can be seen under a light microscope with a PAS stain.[5]
Prognosis follows the rule of thirds: one-third remain with MGN indefinitely, one-third remit, and one-third progress to end-stage kidney failure. As the glomerulonephritis progresses, the tubules of the kidney become infected, leading to atrophy and hyalinisation. The kidney appears to shrink. Treatment with corticosteroids is attempted if the disease progresses.
In extremely rare cases, the disease has been known to run in families, usually passed down through the females. This condition, similarly, is called Familial Membranous Glomerulonephritis. There have only been about nine documented cases in the world.
Thin basement membrane disease
Thin basement membrane disease is an autosomal dominant inherited disease characterized by thin glomerular basement membranes on electron microscopy. It is a benign condition that causes persistent microscopic hematuria. This also may cause proteinuria which is usually mild and overall prognosis is excellent.
Fibronectin glomerulopathy
Fibronectin glomerulopathy is a rare form of glomerulopathy characterised by enlarged glomeruli with deposits in the mesangium and subendothelial space. The deposits have been shown to be fibronectin. This condition is inherited in an autosomal dominant fashion. About 40% of cases are due to mutations in the fibronectin (FN1) gene located on chromosome 2 (2q34).
Proliferative
Proliferative glomerulonephritis is characterised by an increased number of cells in the glomerulus. These forms usually present with a triad of blood in the urine, decreased urine production, and hypertension, the nephritic syndrome. These forms usually progress to end-stage kidney failure (ESKF) over weeks to years (depending on type).
IgA nephropathy
IgA nephropathy, also known as Berger's disease, is the most common type of glomerulonephritis, and generally presents with isolated visible or occult hematuria, occasionally combined with low grade proteinuria, and rarely causes a nephritic syndrome characterised by protein in the urine, and visible blood in the urine. IgA nephropathy is classically described as a self-resolving form in young adults several days after a respiratory infection. It is characterised by deposits of IgA in the space between glomerular capillaries.[2]:501[4]:554–555
Henoch–Schönlein purpura refers to a form of IgA nephropathy, typically affecting children, characterised by a rash of small bruises affecting the buttocks and lower legs, with abdominal pain.[2]:501
Post-infectious
Post-infectious glomerulonephritis can occur after essentially any infection, but classically occurs after infection with the bacteria Streptococcus pyogenes. It typically occurs 1–4 weeks after a pharyngeal infection with this bacterium, and is likely to present with malaise, a slight fever, nausea and a mild nephritic syndrome of moderately increased blood pressure, gross haematuria, and smoky-brown urine. Circulating immune complexes that deposit in the glomerules may lead to an inflammatory reaction.[4] :554–555
Diagnosis may be made on clinical findings or through antistreptolysin O antibodies found in the blood. A biopsy is seldom done, and the disease is likely to self-resolve in children in 1–4 weeks, with a poorer prognosis if adults are affected.[2] :501
Membranoproliferative
Membranoproliferative GN (MPGN), also known as mesangiocapillary glomerulonephritis,[2]:502 is characterised by an increase in the number of cells in the glomerulus, and alterations in the glomerular basement membrane. These forms present with the nephritic syndrome, hypocomplementemia, and have a poor prognosis. Two primary subtypes exist:[4]:552–553
- Type 1 MPGN is caused by circulating immune complexes, typically secondary to systemic lupus erythematosus, hepatitis B and C, or other chronic or recurring infections. Circulating immune complexes may activate the complement system, leading to inflammation and an influx of inflammatory cells.[4]:552–553
- Type 2 MPGN, also known as Dense Deposit Disease, is characterised by an excessive activation of the complement system. The C3 Nephritic Factor autoantibody stabilizes C3-convertase, which may lead to an excessive activation of complement.[4]:553
Rapidly progressive glomerulonephritis
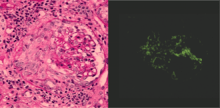
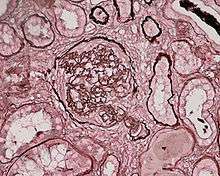
Rapidly progressive glomerulonephritis, also known as crescentic GN, is characterised by a rapid, progressive deterioration in kidney function. People with rapidly progressive glomerulonephritis may present with a nephritic syndrome. In management, steroid therapy is sometimes used, although the prognosis remains poor.[6] Three main subtypes are recognised:[4]:557–558
- Type 1 is Goodpasture syndrome, an autoimmune disease also affecting the lung. In Goodpasture syndrome, IgG antibodies directed against the glomerular basement membrane trigger an inflammatory reaction, causing a nephritic syndrome and the coughing up of blood.[4]:557 High dose immunosuppression is required (intravenous methylprednisolone) and cyclophosphamide, plus plasmapheresis. Immunohistochemistry staining of tissue specimens shows linear IgG deposits.
- Type 2 is characterised by immune-complex-mediated damage, and may be associated with systemic lupus erythematosus, post-infective glomerulonephritis, IgA nephropathy, and IgA vasculitis.[4]:558
- Type 3 rapidly progressive glomerulonephritis, also called pauciimmune type, is associated with causes of vascular inflammation including granulomatosis with polyangiitis (GPA) and microscopic polyangiitis. No immune deposits can be seen on staining, however blood tests may be positive for the ANCA antibody.[4]:558–559
Histopathologically, the majority of glomeruli present "crescents". Formation of crescents is initiated by passage of fibrin into the Bowman space as a result of increased permeability of glomerular basement membrane. Fibrin stimulates the proliferation of endothelial cells of Bowman capsule, and an influx of monocytes. Rapid growing and fibrosis of crescents compresses the capillary loops and decreases the Bowman space, which leads to kidney failure within weeks or months.
Diagnosis
Some forms of glomerulonephritis are diagnosed clinically, based on findings on history and examination. Other tests may include:[6]
- Urine examination
- Blood tests investigating the cause, including FBC, inflammatory markers, and special tests (including ASLO, ANCA, Anti-GBM, Complement levels, Anti-nuclear antibodies)
- Biopsy of the kidney
- Renal ultrasonography is useful for prognostic purposes in finding signs of chronic kidney disease, which however may be caused by many other diseases than glomerulonephritis.[7]
Treatment
Antibiotic therapy to prevent streptococcal infection ( Prophylaxis ). Steroids to suppress immunity. Provide high calories & Low protein, sodium & potassium diet. Monitor for sign of kidney failure, heart failure, and hypertensive encephalopathy.
References
- "glomerulonephritis" at Dorland's Medical Dictionary
- Colledge, Nicki R.; Walker, Brian R.; Ralston, Stuart H., eds. (2010). Davidson's principles and practice of medicine. illust. Robert Britton (21st ed.). Edinburgh: Churchill Livingstone/Elsevier. ISBN 978-0-7020-3084-0.
- The Nephrotic Syndrome Stephan R. Orth, M.D., and Eberhard Ritz, M.D. N Engl J Med 1998; 338:1202–1211 April 23, 1998 DOI: 10.1056/NEJM199804233381707
- Kumar, Vinay, ed. (2007). Robbins basic pathology (8th ed.). Philadelphia: Saunders/Elsevier. ISBN 978-1-4160-2973-1.
- MedlinePlus Encyclopedia Membranous nephropathy
- COUSER, W (1 May 1999). "Glomerulonephritis". The Lancet. 353 (9163): 1509–1515. doi:10.1016/S0140-6736(98)06195-9. PMID 10232333.
- Content initially copied from: Hansen, Kristoffer; Nielsen, Michael; Ewertsen, Caroline (2015). "Ultrasonography of the Kidney: A Pictorial Review". Diagnostics. 6 (1): 2. doi:10.3390/diagnostics6010002. ISSN 2075-4418. PMC 4808817. PMID 26838799. (CC-BY 4.0)
External links
| Classification | |
|---|---|
| External resources |