Streptococcus pyogenes
Streptococcus pyogenes is a species of Gram-positive, aerotolerant bacterium in the genus Streptococcus. These bacteria are extracellular, and made up of non-motile and non-sporing cocci. It is clinically important for humans. It is an infrequent, but usually pathogenic, part of the skin microbiota. It is the predominant species harboring the Lancefield group A antigen, and is often called group A Streptococcus (GAS). However, both Streptococcus dysgalactiae and the Streptococcus anginosus group can possess group A antigen. Group A streptococci when grown on blood agar typically produces small zones of beta-hemolysis, a complete destruction of red blood cells. (A zone size of 2–3 mm is typical.) It is thus also called group A (beta-hemolytic) Streptococcus (GABHS), and it can make colonies greater than 5 mm in size.[1]
| Streptococcus pyogenes | |
|---|---|
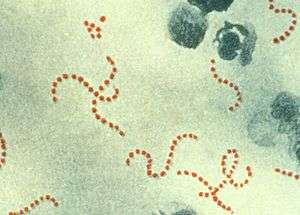 | |
| S. pyogenes bacteria at 900× magnification | |
| Scientific classification | |
| Domain: | Bacteria |
| Phylum: | Firmicutes |
| Class: | Bacilli |
| Order: | Lactobacillales |
| Family: | Streptococcaceae |
| Genus: | Streptococcus |
| Species: | S. pyogenes |
| Binomial name | |
| Streptococcus pyogenes Rosenbach 1884 | |
Like other cocci, streptococci are round bacteria. The species name is derived from Greek words meaning 'a chain' (streptos) of berries (coccus [Latinized from kokkos]) and pus (pyo)-forming(genes), because streptococcal cells tend to link in chains of round cells (see image) and a number of infections caused by the bacterium produce pus. The main criterion for differentiation between Staphylococcus spp. and Streptococcus spp. is the catalase test. Staphylococci are catalase positive whereas streptococci are catalase-negative.[2] S. pyogenes can be cultured on fresh blood agar plates. Under ideal conditions, it has an incubation period of 1 to 3 days.[3]
An estimated 700 million GAS infections occur worldwide each year. While the overall mortality rate for these infections is 0.1%, over 650,000 of the cases are severe and invasive, and have a mortality rate of 25%.[4] Early recognition and treatment are critical; diagnostic failure can result in sepsis and death.[5][6]
Epidemiology
S. pyogenes typically colonizes the throat, genital mucosa, rectum, and skin. Of healthy individuals, 1% to 5% have throat, vaginal, or rectal carriage. In healthy children, such carriage rate varies from 2% to 17%. There are four methods for the transmission of this bacterium: inhalation of respiratory droplets, skin contact, contact with objects, surface, or dust that is contaminated with bacteria or, less commonly, transmission through food. Such bacteria can cause a variety of diseases such as streptococcal pharyngitis, rheumatic fever, rheumatic heart disease, and scarlet fever. Although pharyngitis is mostly viral in origin, about 15 to 30% of all pharyngitis cases in children are caused by GAS; meanwhile, 5 to 20% of pharyngitis in adults are streptococcal. The number of pharyngitis cases is higher in children when compared with adults due to exposures in schools, nurseries, and as a consequence of lower host immunity. Such cases Streptococcal pharyngitis occurs more frequently from December to April (later winter to early spring) in seasonal countries, possibly due to changing climate, behavioural changes or predisposing viral infection. Disease cases are the lowest during autumn.[7]
MT1 (metabolic type 1) clone is frequently associated with invasive Streptococcus pyogenes infections among developed countries. The incidence and mortality of S. pyognes was high during the pre-penicillin era, but had already started to fall prior to the widespread availability of penicillin. Therefore, environmental factors do play a role in the S. pyogenes infection. Incidence of S. pyogenes is 2 to 4 per 100,000 population in developed countries and 12 to 83 per 100,000 population in developing countries. S. pyogenes infection is more frequently found in men than women, with highest rates in the elderly, followed by infants. In people with risk factors such as heart disease, diabetes, malignancy, blunt trauma, surgical incision, virus respiratory infection, including influenza, S. pyogenes infection happens in 17 to 25% of all cases. GAS secondary infection usually happens within one week of the diagnosis of influenza infection. In 14 to 16% of childhood S. pyogenes infections, there is a prior chickenpox infection. Such S. pyogenes infection in children usually manifests as severe soft tissue infection with onset 4 to 12 days from the chickenpox diagnosis. There is also 40 to 60 times increase in risk of S. pyogenes infection within the first two weeks of chickenpox infection in children. However, 20 to 30% of S. pyogenes infection does occur in adults with no identifiable risk factors. The incidence is higher in children (50 to 80% of S. pyogenes infection) with no known risk factors. The rates of scarlet fever in UK was usually 4 in 100,000 population, however, in 2014, the rates had risen to 49 per 100,000 population. Rheumatic fever and rheumatic heart disease (RHD) usually occurs at 2 to 3 weeks after the throat infection, which is more common among the impoverished people in developing countries. From 1967 to 1996, the global mean incidence of rheumatic fever and RHD was 19 per 100,000 with the highest incidence at 51 per 100,000.[7]
Maternal S. pyogenes infection usually happens in late pregnancy; at more than 30 weeks of gestation to four weeks post partum, which accounts for 2 to 4% of all the S. pyogenes infections. This represents 20 to 100 times increase in risk for S. pyogenes infections. Clinical manifestations are: pneumonia, septic arthritis, necrotizing fasciitis, and genital tract sepsis. According to a study done by Queen Charlotte's hospital in London during the 1930s, the vagina was not the common source of such infection. On the contrary, maternal throat infection and close contacts with carriers were the more common sites for maternal S. pyogenes infection.[7]
Bacteriology
Serotyping
In 1928, Rebecca Lancefield published a method for serotyping S. pyogenes based on its cell-wall polysaccharide,[8] a virulence factor displayed on its surface.[9] Later, in 1946, Lancefield described the serologic classification of S. pyogenes isolates based on their surface T-antigen.[10] Four of the 20 T-antigens have been revealed to be pili, which are used by bacteria to attach to host cells.[11] As of 2016, a total of 120 M proteins are identified. These M proteins are encoded by 234 types emm gene with greater than 1,200 alleles.[7]
Lysogeny
All strains of S. pyogenes are polylysogenized, in that they carry one or more bacteriophage on their genomes.[12] Some of the 'phages may be defective, but in some cases active 'phage may compensate for defects in others.[13] In general, the genome of S. pyogenes strains isolated during disease are >90% identical, they differ by the 'phage they carry.[14]
Virulence factors
S. pyogenes has several virulence factors that enable it to attach to host tissues, evade the immune response, and spread by penetrating host tissue layers.[15] A carbohydrate-based bacterial capsule composed of hyaluronic acid surrounds the bacterium, protecting it from phagocytosis by neutrophils.[2] In addition, the capsule and several factors embedded in the cell wall, including M protein, lipoteichoic acid, and protein F (SfbI) facilitate attachment to various host cells.[16] M protein also inhibits opsonization by the alternative complement pathway by binding to host complement regulators. The M protein found on some serotypes is also able to prevent opsonization by binding to fibrinogen.[2] However, the M protein is also the weakest point in this pathogen's defense, as antibodies produced by the immune system against M protein target the bacteria for engulfment by phagocytes. M proteins are unique to each strain, and identification can be used clinically to confirm the strain causing an infection.[17]
| Name | Description |
|---|---|
| Streptolysin O | An exotoxin, one of the bases of the organism's beta-hemolytic property, streptolysin O causes an immune response and detection of antibodies to it; antistreptolysin O (ASO) can be clinically used to confirm a recent infection. It is damaged by oxygen. |
| Streptolysin S | A cardiotoxic exotoxin, another beta-hemolytic component, not immunogenic and O2 stable: A potent cell poison affecting many types of cell including neutrophils, platelets, and subcellular organelles. |
| Streptococcal pyrogenic exotoxin A (SpeA) | Superantigens secreted by many strains of S. pyogenes: This pyrogenic exotoxin is responsible for the rash of scarlet fever and many of the symptoms of streptococcal toxic shock syndrome, also known as toxic shock like syndrome(TSLS). |
| Streptococcal pyrogenic exotoxin C (SpeC) | |
| Streptokinase | Enzymatically activates plasminogen, a proteolytic enzyme, into plasmin, which in turn digests fibrin and other proteins |
| Hyaluronidase | Hyaluronidase is widely assumed to facilitate the spread of the bacteria through tissues by breaking down hyaluronic acid, an important component of connective tissue. However, very few isolates of S. pyogenes are capable of secreting active hyaluronidase due to mutations in the gene that encode the enzyme. Moreover, the few isolates capable of secreting hyaluronidase do not appear to need it to spread through tissues or to cause skin lesions.[18] Thus, the true role of hyaluronidase in pathogenesis, if any, remains unknown. |
| Streptodornase | Most strains of S. pyogenes secrete up to four different DNases, which are sometimes called streptodornase. The DNases protect the bacteria from being trapped in neutrophil extracellular traps (NETs) by digesting the NETs' web of DNA, to which are bound neutrophil serine proteases that can kill the bacteria.[19] |
| C5a peptidase | C5a peptidase cleaves a potent neutrophil chemotaxin called C5a, which is produced by the complement system.[20] C5a peptidase is necessary to minimize the influx of neutrophils early in infection as the bacteria are attempting to colonize the host's tissue.[21] C5a peptidase, although required to degrade the neutrophil chemotaxin C5a in the early stages of infection, is not required for S. pyogenes to prevent the influx of neutrophils as the bacteria spread through the fascia.[22] |
| Streptococcal chemokine protease | The affected tissue of patients with severe cases of necrotizing fasciitis are devoid of neutrophils.[23] The serine protease ScpC, which is released by S. pyogenes, is responsible for preventing the migration of neutrophils to the spreading infection. ScpC degrades the chemokine IL-8, which would otherwise attract neutrophils to the site of infection.[21][22] |
Disease
S. pyogenes is the cause of many human diseases, ranging from mild superficial skin infections to life-threatening systemic diseases.[2] Infections typically begin in the throat or skin. The most striking sign is a strawberry-like rash. Examples of mild S. pyogenes infections include pharyngitis (strep throat) and localized skin infection (impetigo). Erysipelas and cellulitis are characterized by multiplication and lateral spread of S. pyogenes in deep layers of the skin. S. pyogenes invasion and multiplication in the fascia can lead to necrotizing fasciitis, a life-threatening condition requiring surgery. [29] The bacterium is found in neonatal infections.[30]
Infections due to certain strains of S. pyogenes can be associated with the release of bacterial toxins. Throat infections associated with release of certain toxins lead to scarlet fever. Other toxigenic S. pyogenes infections may lead to streptococcal toxic shock syndrome, which can be life-threatening.[2]
S. pyogenes can also cause disease in the form of post-infectious "non-pyogenic" (not associated with local bacterial multiplication and pus formation) syndromes. These autoimmune-mediated complications follow a small percentage of infections and include rheumatic fever and acute post-infectious glomerulonephritis. Both conditions appear several weeks following the initial streptococcal infection. Rheumatic fever is characterized by inflammation of the joints and/or heart following an episode of streptococcal pharyngitis. Acute glomerulonephritis, inflammation of the renal glomerulus, can follow streptococcal pharyngitis or skin infection.
This bacterium remains acutely sensitive to penicillin. Failure of treatment with penicillin is generally attributed to other local commensal organisms producing β-lactamase, or failure to achieve adequate tissue levels in the pharynx. Certain strains have developed resistance to macrolides, tetracyclines, and clindamycin.
Applications
See also
- Friedrich Fehleisen
- Friedrich Julius Rosenbach
- Friedrich Loeffler
- Frederick Twort
References
- "Streptococcus pyogenes - Pathogen Safety Data Sheets". Government of Canada, Public Health Agency of Canada. 2001-09-26.
- Ryan KJ, Ray CG, eds. (2004). Sherris Medical Microbiology (4th ed.). McGraw Hill. ISBN 978-0-8385-8529-0.
- Streptococcal Pharyngitis, archived from the original on May 13, 2012
- Aziz RK, Kansal R, Aronow BJ, Taylor WL, Rowe SL, Kubal M, Chhatwal GS, Walker MJ, Kotb M (2010). Ahmed N (ed.). "Microevolution of Group A Streptococci In Vivo: Capturing Regulatory Networks Engaged in Sociomicrobiology, Niche Adaptation, and Hypervirulence". PLoS ONE. 5 (4): e9798. Bibcode:2010PLoSO...5.9798A. doi:10.1371/journal.pone.0009798. PMC 2854683. PMID 20418946.
- Jim Dwyer (July 11, 2012). "An Infection, Unnoticed, Turns Unstoppable". The New York Times. Retrieved July 12, 2012.
- Jim Dwyer (July 18, 2012). "After Boy's Death, Hospital Alters Discharging Procedures". The New York Times. Retrieved July 19, 2012.
- Androulla, Efstratiou; Theresa, Lamagni (10 February 2016). "Epidemiology of Streptococcus pyogenes". Streptococcus pyogenes : Basic Biology to Clinical Manifestations. Oklahoma City, United States: University of Oklahoma Health Sciences Center. Retrieved 24 February 2018.
- Pignanelli S, Brusa S, Pulcrano G, Catania MR, Cocchi E, Lanari M (2015). "A rare case of infant sepsis due to the emm-89 genotype of Group A Streptococcus within a community-acquired cluster". New Microbiol. 38 (4): 589–92. PMID 26485019.
- Lancefield RC (1928). "The antigenic complex of Streptococcus hemolyticus". J Exp Med. 47 (1): 9–10. doi:10.1084/jem.47.1.91. PMC 2131344. PMID 19869404.
- Lancefield RC, Dole VP (1946). "The properties of T antigen extracted from group A hemolytic streptococci". J Exp Med. 84 (5): 449–71. doi:10.1084/jem.84.5.449. PMC 2135665. PMID 19871581.
- Mora M, Bensi G, Capo S, Falugi F, Zingaretti C, Manetti AG, Maggi T, Taddei AR, Grandi G, Telford JL (2005). "Group A Streptococcus produce pilus-like structures containing protective antigens and Lancefield T antigens". Proc Natl Acad Sci USA. 102 (43): 15641–6. Bibcode:2005PNAS..10215641M. doi:10.1073/pnas.0507808102. PMC 1253647. PMID 16223875.
- Ferretti JJ; McShan WM; Ajdic D; Savic DJ; Savic G; Lyon K; et al. (2001). "Complete Genome Sequence of an M1 Strain of Streptococcus pyogenes". Proc Natl Acad Sci USA. 98 (8): 4658–63. Bibcode:2001PNAS...98.4658F. doi:10.1073/pnas.071559398. PMC 31890. PMID 11296296.
- Canchaya C, Desiere F, McShan WM, Ferretti JJ, Parkhill J, Brussow H (2002). "Genome analysis of an inducible prophage and prophage remnants integrated in the Streptococcus pyogenes strain SF370". Virology. 302 (2): 245–58. doi:10.1006/viro.2002.1570. PMID 12441069.
- Banks DJ, Porcella SF, Barbian KD, Martin JM, Musser JM (2003). "Structure and distribution of an unusual chimeric genetic element encoding macrolide resistance in phylogenetically diverse clones of group A Streptococcus". J Infect Dis. 188 (12): 1898–908. doi:10.1086/379897. PMID 14673771.
- Patterson MJ (1996). "Streptococcus". In Baron S; et al. (eds.). Streptococcus. In: Baron's Medical Microbiology (4th ed.). Univ of Texas Medical Branch. ISBN 978-0-9631172-1-2.
- Bisno AL, Brito MO, Collins CM (2003). "Molecular basis of group A streptococcal virulence". Lancet Infect Dis. 3 (4): 191–200. doi:10.1016/S1473-3099(03)00576-0. PMID 12679262.
- Engel ME, Muhamed B, Whitelaw AC, Musvosvi M, Mayosi BM, Dale JB. Group A streptococcal emm type prevalence among symptomatic children in Cape Town and potential vaccine coverage. Pediatr Infect Dis J. 2014 Feb;33(2):208-10. doi: 10.1097/INF.0b013e3182a5c32a.PMID 23934204
- Starr CR, Engleberg NC (2006). "Role of Hyaluronidase in Subcutaneous Spread and Growth of Group A Streptococcus". Infect Immun. 74 (1): 40–8. doi:10.1128/IAI.74.1.40-48.2006. PMC 1346594. PMID 16368955.
- Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V (2006). "DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps" (PDF). Curr Biol. 16 (4): 396–400. doi:10.1016/j.cub.2005.12.039. PMID 16488874.
- Wexler DE, Chenoweth DE, Cleary PP (1985). "Mechanism of action of the group A streptococcal C5a inactivator". Proc Natl Acad Sci USA. 82 (23): 8144–8. Bibcode:1985PNAS...82.8144W. doi:10.1073/pnas.82.23.8144. PMC 391459. PMID 3906656.
- Ji Y, McLandsborough L, Kondagunta A, Cleary PP (1996). "C5a peptidase alters clearance and trafficking of group A streptococci by infected mice". Infect Immun. 64 (2): 503–10. PMC 173793. PMID 8550199.
- Hidalgo-Grass C, Mishalian I, Dan-Goor M, Belotserkovsky I, Eran Y, Nizet V, Peled A, Hanski E (2006). "A streptococcal protease that degrades CXC chemokines and impairs bacterial clearance from infected tissues". EMBO J. 25 (19): 4628–37. doi:10.1038/sj.emboj.7601327. PMC 1589981. PMID 16977314.
- Hidalgo-Grass C, Dan-Goor M, Maly A, Eran Y, Kwinn LA, Nizet V, Ravins M, Jaffe J, Peyser A, Moses AE, Hanski E (2004). "Effect of a bacterial pheromone peptide on host chemokine degradation in group A streptococcal necrotising soft-tissue infections". Lancet. 363 (9410): 696–703. doi:10.1016/S0140-6736(04)15643-2. PMID 15001327.
- Beres SB, Richter EW, Nagiec MJ, Sumby P, Porcella SF, DeLeo FR, Musser JM (2006). "Molecular genetic anatomy of inter- and intraserotype variation in the human bacterial pathogen group a Streptococcus". Proceedings of the National Academy of Sciences. 103 (18): 7059–64. Bibcode:2006PNAS..103.7059B. doi:10.1073/pnas.0510279103. PMC 1459018. PMID 16636287.
- "Streptococcus pyogenes NZ131".
- McShan, W. M.; Ferretti, J. J.; Karasawa, T; Suvorov, A. N.; Lin, S; Qin, B; Jia, H; Kenton, S; Najar, F; Wu, H; Scott, J; Roe, B. A.; Savic, D. J. (2008). "Genome sequence of a nephritogenic and highly transformable M49 strain of Streptococcus pyogenes". Journal of Bacteriology. 190 (23): 7773–85. doi:10.1128/JB.00672-08. PMC 2583620. PMID 18820018.
- Sumby, P; Porcella, S. F.; Madrigal, A. G.; Barbian, K. D.; Virtaneva, K; Ricklefs, S. M.; Sturdevant, D. E.; Graham, M. R.; Vuopio-Varkila, J; Hoe, N. P.; Musser, J. M. (2005). "Evolutionary origin and emergence of a highly successful clone of serotype M1 group a Streptococcus involved multiple horizontal gene transfer events". The Journal of Infectious Diseases. 192 (5): 771–82. doi:10.1086/432514. PMID 16088826.
- "Streptococcus pyogenes MGAS5005".
- "Necrotizing Fasciitis". CDC. Content source: National Center for Immunization and Respiratory Diseases, Division of Bacterial Diseases. Page maintained by: Office of the Associate Director for Communication, Digital Media Branch, Division of Public Affairs. October 26, 2017. Retrieved 2018-01-06.
- Baucells, B.J.; Mercadal Hally, M.; Álvarez Sánchez, A.T.; Figueras Aloy, J. (2015). "Asociaciones de probióticos para la prevención de la enterocolitis necrosante y la reducción de la sepsis tardía y la mortalidad neonatal en recién nacidos pretérmino de menos de 1.500g: una revisión sistemática". Anales de Pediatría. 85 (5): 247–255. doi:10.1016/j.anpedi.2015.07.038. ISSN 1695-4033. PMID 26611880.
- "Flesh-eating bacteria inspire superglue - University of Oxford".
- Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M (2012). "Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin". Proceedings of the National Academy of Sciences. 109 (12): E690–7. doi:10.1073/pnas.1115485109. PMC 3311370. PMID 22366317.
- Baruah K, Bowden TA, Krishna BA, Dwek RA, Crispin M, Scanlan CN (2012). "Selective Deactivation of Serum IgG: A General Strategy for the Enhancement of Monoclonal Antibody Receptor Interactions". Journal of Molecular Biology. 420 (1–2): 1–7. doi:10.1016/j.jmb.2012.04.002. PMC 3437440. PMID 22484364.
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E (March 2011). "CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III". Nature. 471 (7340): 602–607. doi:10.1038/nature09886. PMC 3070239. PMID 21455174.
- Zimmer, Carl (2016-06-03). "Scientists Find Form of Crispr Gene Editing With New Capabilities". The New York Times. ISSN 0362-4331. Retrieved 2016-06-10.
Further reading
- Freiberg JA, McIver KS, Shirtliff ME (2014). "In vivo expression of Streptococcus pyogenes immunogenic proteins during tibial foreign body infection". Infect. Immun. 82 (9): 3891–9. doi:10.1128/IAI.01831-14. PMC 4187806. PMID 25001603.
- Rosenbach FJ (1884). Mikro-Organismen bei den Wund-Infections-Krankheiten des Menschen (in German). J.F. Bergmann. OL 22886502M.
- Wilson LG (October 1987). "The early recognition of streptococci as causes of disease". Med Hist. 31 (4): 403–14. doi:10.1017/s0025727300047268. PMC 1139783. PMID 3316876.
- Rolleston JD (November 1928). "The history of scarlet fever". British Medical Journal. 2 (3542): 926–9. doi:10.1136/bmj.2.3542.926. PMC 2456687. PMID 20774279.
- World Health Organization (2005). "The current evidence for the burden of group A streptococcal diseases" (PDF). Retrieved 2011-08-22.
- Carapetis JR, Steer AC, Mulholland EK, Weber M (November 2005). "The global burden of group A streptococcal diseases". Lancet Infect Dis. 5 (11): 685–94. doi:10.1016/S1473-3099(05)70267-X. PMID 16253886. (corresponding summary article)
- Ferretti JJ, Stevens DL, Fischetti VA, eds. (2016). Streptococcus pyogenes: Basic Biology to Clinical Manifestations [Internet]. Oklahoma City, OK: University of Oklahoma Health Sciences Center.