Antibody
An antibody (Ab), also known as an immunoglobulin (Ig),[1] is a large, Y-shaped protein produced mainly by plasma cells that is used by the immune system to neutralize pathogens such as pathogenic bacteria and viruses. The antibody recognizes a unique molecule of the pathogen, called an antigen, via the fragment antigen-binding (Fab) variable region.[2][3] Each tip of the "Y" of an antibody contains a paratope (analogous to a lock) that is specific for one particular epitope it is place where (similarly, analogous to a key) on an antigen, allowing these two structures to bind together with precision. Using this binding mechanism, an antibody can tag a microbe or an infected cell for attack by other parts of the immune system, or can neutralize its target directly (for example, by inhibiting a part of a microbe that is essential for its invasion and survival). Depending on the antigen, the binding may impede the biological process causing the disease or may activate macrophages to destroy the foreign substance. The ability of an antibody to communicate with the other components of the immune system is mediated via its Fc region (located at the base of the "Y"), which contains a conserved glycosylation site involved in these interactions.[4] The production of antibodies is the main function of the humoral immune system.[5]
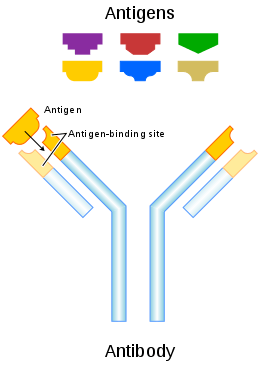
Antibodies are secreted by B cells of the adaptive immune system, mostly by differentiated B cells called plasma cells. Antibodies can occur in two physical forms, a soluble form that is secreted from the cell to be free in the blood plasma, and a membrane-bound form that is attached to the surface of a B cell and is referred to as the B-cell receptor (BCR). The BCR is found only on the surface of B cells and facilitates the activation of these cells and their subsequent differentiation into either antibody factories called plasma cells or memory B cells that will survive in the body and remember that same antigen so the B cells can respond faster upon future exposure.[6] In most cases, interaction of the B cell with a T helper cell is necessary to produce full activation of the B cell and, therefore, antibody generation following antigen binding.[7] Soluble antibodies are released into the blood and tissue fluids, as well as many secretions to continue to survey for invading microorganisms.
Antibodies are glycoproteins belonging to the immunoglobulin superfamily.[4] They constitute most of the gamma globulin fraction of the blood proteins. They are typically made of basic structural units—each with two large heavy chains and two small light chains. There are several different types of antibody heavy chains that define the five different types of crystallisable fragments (Fc) that may be attached to the antigen-binding fragments. The five different types of Fc regions allow antibodies to be grouped into five isotypes. Each Fc region of a particular antibody isotype is able to bind to its specific Fc Receptor (except for IgD, which is essentially the BCR), thus allowing the antigen-antibody complex to mediate different roles depending on which FcR it binds. The ability of an antibody to bind to its corresponding FcR is further modulated by the structure of the glycan(s) present at conserved sites within its Fc region.[4] The ability of antibodies to bind to FcRs helps to direct the appropriate immune response for each different type of foreign object they encounter.[8] For example, IgE is responsible for an allergic response consisting of mast cell degranulation and histamine release. IgE's Fab paratope binds to allergic antigen, for example house dust mite particles, while its Fc region binds to Fc receptor ε. The allergen-IgE-FcRε interaction mediates allergic signal transduction to induce conditions such as asthma.[9]
Though the general structure of all antibodies is very similar, a small region at the tip of the protein is extremely variable, allowing millions of antibodies with slightly different tip structures, or antigen-binding sites, to exist. This region is known as the hypervariable region. Each of these variants can bind to a different antigen.[2] This enormous diversity of antibody paratopes on the antigen-binding fragments allows the immune system to recognize an equally wide variety of antigens.[1] The large and diverse population of antibody paratope is generated by random recombination events of a set of gene segments that encode different antigen-binding sites (or paratopes), followed by random mutations in this area of the antibody gene, which create further diversity.[8][10] This recombinational process that produces clonal antibody paratope diversity is called V(D)J or VJ recombination. Basically, the antibody paratope is polygenic, made up of three genes, V, D, and J. Each paratope locus is also polymorphic, such that during antibody production, one allele of V, one of D, and one of J is chosen. These gene segments are then joined together using random genetic recombination to produce the paratope. The regions where the genes are randomly recombined together is the hyper variable region used to recognise different antigens on a clonal basis.
Antibody genes also re-organize in a process called class switching that changes the one type of heavy chain Fc fragment to another, creating a different isotype of the antibody that retains the antigen-specific variable region. This allows a single antibody to be used by different types of Fc receptors, expressed on different parts of the immune system.
History
The first use of the term "antibody" occurred in a text by Paul Ehrlich. The term Antikörper (the German word for antibody) appears in the conclusion of his article "Experimental Studies on Immunity", published in October 1891, which states that, "if two substances give rise to two different Antikörper, then they themselves must be different".[11] However, the term was not accepted immediately and several other terms for antibody were proposed; these included Immunkörper, Amboceptor, Zwischenkörper, substance sensibilisatrice, copula, Desmon, philocytase, fixateur, and Immunisin.[11] The word antibody has formal analogy to the word antitoxin and a similar concept to Immunkörper (immune body in English).[11] As such, the original construction of the word contains a logical flaw; the antitoxin is something directed against a toxin, while the antibody is a body directed against something.[11]

The study of antibodies began in 1890 when Kitasato Shibasaburō described antibody activity against diphtheria and tetanus toxins. Kitasato put forward the theory of humoral immunity, proposing that a mediator in serum could react with a foreign antigen.[15][16] His idea prompted Paul Ehrlich to propose the side-chain theory for antibody and antigen interaction in 1897, when he hypothesized that receptors (described as "side-chains") on the surface of cells could bind specifically to toxins – in a "lock-and-key" interaction – and that this binding reaction is the trigger for the production of antibodies.[17] Other researchers believed that antibodies existed freely in the blood and, in 1904, Almroth Wright suggested that soluble antibodies coated bacteria to label them for phagocytosis and killing; a process that he named opsoninization.[18]

In the 1920s, Michael Heidelberger and Oswald Avery observed that antigens could be precipitated by antibodies and went on to show that antibodies are made of protein.[19] The biochemical properties of antigen-antibody-binding interactions were examined in more detail in the late 1930s by John Marrack.[20] The next major advance was in the 1940s, when Linus Pauling confirmed the lock-and-key theory proposed by Ehrlich by showing that the interactions between antibodies and antigens depend more on their shape than their chemical composition.[21] In 1948, Astrid Fagreaus discovered that B cells, in the form of plasma cells, were responsible for generating antibodies.[22]
Further work concentrated on characterizing the structures of the antibody proteins. A major advance in these structural studies was the discovery in the early 1960s by Gerald Edelman and Joseph Gally of the antibody light chain,[23] and their realization that this protein is the same as the Bence-Jones protein described in 1845 by Henry Bence Jones.[24] Edelman went on to discover that antibodies are composed of disulfide bond-linked heavy and light chains. Around the same time, antibody-binding (Fab) and antibody tail (Fc) regions of IgG were characterized by Rodney Porter.[25] Together, these scientists deduced the structure and complete amino acid sequence of IgG, a feat for which they were jointly awarded the 1972 Nobel Prize in Physiology or Medicine.[25] The Fv fragment was prepared and characterized by David Givol.[26] While most of these early studies focused on IgM and IgG, other immunoglobulin isotypes were identified in the 1960s: Thomas Tomasi discovered secretory antibody (IgA);[27] David S. Rowe and John L. Fahey discovered IgD;[28] and Kimishige Ishizaka and Teruko Ishizaka discovered IgE and showed it was a class of antibodies involved in allergic reactions.[29] In a landmark series of experiments beginning in 1976, Susumu Tonegawa showed that genetic material can rearrange itself to form the vast array of available antibodies.[30]
Forms
The membrane-bound form of an antibody may be called a surface immunoglobulin (sIg) or a membrane immunoglobulin (mIg). It is part of the B cell receptor (BCR), which allows a B cell to detect when a specific antigen is present in the body and triggers B cell activation.[7] The BCR is composed of surface-bound IgD or IgM antibodies and associated Ig-α and Ig-β heterodimers, which are capable of signal transduction.[31] A typical human B cell will have 50,000 to 100,000 antibodies bound to its surface.[31] Upon antigen binding, they cluster in large patches, which can exceed 1 micrometer in diameter, on lipid rafts that isolate the BCRs from most other cell signaling receptors.[31] These patches may improve the efficiency of the cellular immune response.[32] In humans, the cell surface is bare around the B cell receptors for several hundred nanometers,[31] which further isolates the BCRs from competing influences.
Antibody–antigen interactions
The antibody's paratope interacts with the antigen's epitope. An antigen usually contains different epitopes along its surface arranged discontinuously, and dominant epitopes on a given antigen are called determinants.
Antibody and antigen interact by spatial complementarity (lock and key). The molecular forces involved in the Fab-epitope interaction are weak and non-specific – for example electrostatic forces, hydrogen bonds, hydrophobic interactions, and van der Waals forces. This means binding between antibody and antigen is reversible, and the antibody's affinity towards an antigen is relative rather than absolute. Relatively weak binding also means it is possible for an antibody to cross-react with different antigens of different relative affinities.
Often, once an antibody and antigen bind, they become an immune complex, which functions as a unitary object and can act as an antigen in its own right, being countered by other antibodies. Similarly, haptens are small molecules that provoke no immune response by themselves, but once they bind to proteins, the resulting complex or hapten-carrier adduct is antigenic.
Isotypes
Antibodies can come in different varieties known as isotypes or classes. In placental mammals there are five antibody isotypes known as IgA, IgD, IgE, IgG, and IgM. They are each named with an "Ig" prefix that stands for immunoglobulin (a name sometimes used interchangeably with antibody) and differ in their biological properties, functional locations and ability to deal with different antigens, as depicted in the table.[33] The different suffixes of the antibody isotypes denote the different types of heavy chains the antibody contains, with each heavy chain class named alphabetically: α (alpha), γ (gamma), δ (delta), ε (epsilon), and μ (mu). This gives rise to IgA, IgG, IgD, IgE, and IgM, respectively.
| Class | Subclass | Description | Antibody complexes |
|---|---|---|---|
| IgA | 2 | Found in mucosal areas, such as the gut, respiratory tract and urogenital tract, and prevents colonization by pathogens.[34] Also found in saliva, tears, and breast milk. | 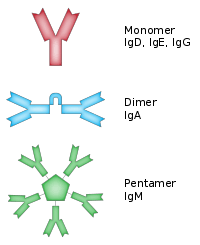 |
| IgD | 1 | Functions mainly as an antigen receptor on B cells that have not been exposed to antigens.[35] It has been shown to activate basophils and mast cells to produce antimicrobial factors.[36] | |
| IgE | 1 | Binds to allergens and triggers histamine release from mast cells and basophils, and is involved in allergy. Also protects against parasitic worms.[5] | |
| IgG | 4 | In its four forms, provides the majority of antibody-based immunity against invading pathogens.[5] The only antibody capable of crossing the placenta to give passive immunity to the fetus. | |
| IgM | 1 | Expressed on the surface of B cells (monomer) and in a secreted form (pentamer) with very high avidity. Eliminates pathogens in the early stages of B cell-mediated (humoral) immunity before there is sufficient IgG.[5][35] |
The antibody isotype of a B cell changes during cell development and activation. Immature B cells, which have never been exposed to an antigen, express only the IgM isotype in a cell surface bound form. The B lymphocyte, in this ready-to-respond form, is known as a "naive B lymphocyte." The naive B lymphocyte expresses both surface IgM and IgD. The co-expression of both of these immunoglobulin isotypes renders the B cell ready to respond to antigen.[37] B cell activation follows engagement of the cell-bound antibody molecule with an antigen, causing the cell to divide and differentiate into an antibody-producing cell called a plasma cell. In this activated form, the B cell starts to produce antibody in a secreted form rather than a membrane-bound form. Some daughter cells of the activated B cells undergo isotype switching, a mechanism that causes the production of antibodies to change from IgM or IgD to the other antibody isotypes, IgE, IgA, or IgG, that have defined roles in the immune system.
| Class | Types | Description |
|---|---|---|
| IgY | Found in birds and reptiles; related to mammalian IgG.[38] | |
| IgW | Found in sharks and skates; related to mammalian IgD.[39] |
Structure
Antibodies are heavy (~150 kDa) globular plasma proteins. The size of an antibody molecule is about 10 nm.[40] They have sugar chains (glycans) added to conserved amino acid residues.[4][41] In other words, antibodies are glycoproteins.[4] The attached glycans are critically important to the structure and function of the antibody.[4] Among other things the expressed glycans can modulate an antibody's affinity for its corresponding FcR(s).[4]
The basic functional unit of each antibody is an immunoglobulin (Ig) monomer (containing only one Ig unit); secreted antibodies can also be dimeric with two Ig units as with IgA, tetrameric with four Ig units like teleost fish IgM, or pentameric with five Ig units, like mammalian IgM.[42]
The variable parts of an antibody are its V regions, and the constant part is its C region.
Immunoglobulin domains
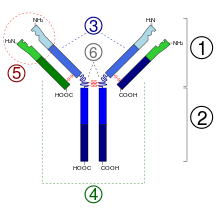
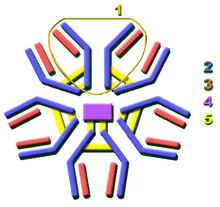
The Ig monomer is a "Y"-shaped molecule that consists of four polypeptide chains; two identical heavy chains and two identical light chains connected by disulfide bonds.[33] Each chain is composed of structural domains called immunoglobulin domains. These domains contain about 70–110 amino acids and are classified into different categories (for example, variable or IgV, and constant or IgC) according to their size and function.[43] They have a characteristic immunoglobulin fold in which two beta sheets create a "sandwich" shape, held together by interactions between conserved cysteines and other charged amino acids.
Heavy chain
There are five types of mammalian Ig heavy chain denoted by the Greek letters: α, δ, ε, γ, and μ.[2] The type of heavy chain present defines the class of antibody; these chains are found in IgA, IgD, IgE, IgG, and IgM antibodies, respectively.[1] Distinct heavy chains differ in size and composition; α and γ contain approximately 450 amino acids, whereas μ and ε have approximately 550 amino acids.[2]
- Fab region
- Fc region
- Heavy chain (blue) with one variable (VH) domain followed by a constant domain (CH1), a hinge region, and two more constant (CH2 and CH3) domains
- Light chain (green) with one variable (VL) and one constant (CL) domain
- Antigen binding site (paratope)
- Hinge regions
Each heavy chain has two regions, the constant region and the variable region. The constant region is identical in all antibodies of the same isotype, but differs in antibodies of different isotypes. Heavy chains γ, α and δ have a constant region composed of three tandem (in a line) Ig domains, and a hinge region for added flexibility;[33] heavy chains μ and ε have a constant region composed of four immunoglobulin domains.[2] The variable region of the heavy chain differs in antibodies produced by different B cells, but is the same for all antibodies produced by a single B cell or B cell clone. The variable region of each heavy chain is approximately 110 amino acids long and is composed of a single Ig domain.
Light chain
In mammals there are two types of immunoglobulin light chain, which are called lambda (λ) and kappa (κ).[2] A light chain has two successive domains: one constant domain and one variable domain. The approximate length of a light chain is 211 to 217 amino acids.[2] Each antibody contains two light chains that are always identical; only one type of light chain, κ or λ, is present per antibody in mammals. Other types of light chains, such as the iota (ι) chain, are found in other vertebrates like sharks (Chondrichthyes) and bony fishes (Teleostei).
CDRs, Fv, Fab and Fc regions
Different parts of an antibody have different functions. Specifically, the "arms" (which are generally identical) contain sites that can bind to specific molecules, enabling recognition of specific antigens. This region of the antibody is called the Fab (fragment, antigen-binding) region. It is composed of one constant and one variable domain from each heavy and light chain of the antibody.[44] The paratope at the amino terminal end of the antibody monomer is shaped by the variable domains from the heavy and light chains. The variable domain is also referred to as the FV region and is the most important region for binding to antigens. To be specific, variable loops of β-strands, three each on the light (VL) and heavy (VH) chains are responsible for binding to the antigen. These loops are referred to as the complementarity determining regions (CDRs). The structures of these CDRs have been clustered and classified by Chothia et al.[45] and more recently by North et al.[46] and Nikoloudis et al.[47] In the framework of the immune network theory, CDRs are also called idiotypes. According to immune network theory, the adaptive immune system is regulated by interactions between idiotypes.
The base of the Y plays a role in modulating immune cell activity. This region is called the Fc (Fragment, crystallizable) region, and is composed of two heavy chains that contribute two or three constant domains depending on the class of the antibody.[2] Thus, the Fc region ensures that each antibody generates an appropriate immune response for a given antigen, by binding to a specific class of Fc receptors, and other immune molecules, such as complement proteins. By doing this, it mediates different physiological effects, including recognition of opsonized particles (binding to FcγR), lysis of cells (binding to complement), and degranulation of mast cells, basophils, and eosinophils (binding to FcεR).[33][48]
In summary, the Fab region of the antibody determines antigen specificity while the Fc region of the antibody determines the antibody's class effect. Since only the constant domains of the heavy chains make up the Fc region of an antibody, the classes of heavy chain in antibodies determine their class effects. Possible classes of heavy chains in antibodies include alpha, gamma, delta, epsilon, and mu, and they define the antibody's isotypes IgA, G, D, E, and M, respectively. This infers different isotypes of antibodies have different class effects due to their different Fc regions binding and activating different types of receptors. Possible class effects of antibodies include: Opsonisation, agglutination, haemolysis, complement activation, mast cell degranulation, and neutralisation (though this class effect may be mediated by the Fab region rather than the Fc region). It also implies that Fab-mediated effects are directed at microbes or toxins, whilst Fc mediated effects are directed at effector cells or effector molecules (see below).
Function
The main categories of antibody action include the following:
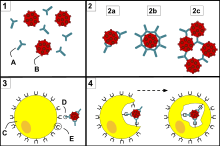
- Neutralisation, in which neutralizing antibodies block parts of the surface of a bacterial cell or virion to render its attack ineffective
- Agglutination, in which antibodies "glue together" foreign cells into clumps that are attractive targets for phagocytosis
- Precipitation, in which antibodies "glue together" serum-soluble antigens, forcing them to precipitate out of solution in clumps that are attractive targets for phagocytosis
- Complement activation (fixation), in which antibodies that are latched onto a foreign cell encourage complement to attack it with a membrane attack complex, which leads to the following:
- Lysis of the foreign cell
- Encouragement of inflammation by chemotactically attracting inflammatory cells
Activated B cells differentiate into either antibody-producing cells called plasma cells that secrete soluble antibody or memory cells that survive in the body for years afterward in order to allow the immune system to remember an antigen and respond faster upon future exposures.[6]
At the prenatal and neonatal stages of life, the presence of antibodies is provided by passive immunization from the mother. Early endogenous antibody production varies for different kinds of antibodies, and usually appear within the first years of life. Since antibodies exist freely in the bloodstream, they are said to be part of the humoral immune system. Circulating antibodies are produced by clonal B cells that specifically respond to only one antigen (an example is a virus capsid protein fragment). Antibodies contribute to immunity in three ways: They prevent pathogens from entering or damaging cells by binding to them; they stimulate removal of pathogens by macrophages and other cells by coating the pathogen; and they trigger destruction of pathogens by stimulating other immune responses such as the complement pathway.[49] Antibodies will also trigger vasoactive amine degranulation to contribute to immunity against certain types of antigens (helminths, allergens).
Activation of complement
Antibodies that bind to surface antigens (for example, on bacteria) will attract the first component of the complement cascade with their Fc region and initiate activation of the "classical" complement system.[49] This results in the killing of bacteria in two ways.[5] First, the binding of the antibody and complement molecules marks the microbe for ingestion by phagocytes in a process called opsonization; these phagocytes are attracted by certain complement molecules generated in the complement cascade. Second, some complement system components form a membrane attack complex to assist antibodies to kill the bacterium directly (bacteriolysis).[50]
Activation of effector cells
To combat pathogens that replicate outside cells, antibodies bind to pathogens to link them together, causing them to agglutinate. Since an antibody has at least two paratopes, it can bind more than one antigen by binding identical epitopes carried on the surfaces of these antigens. By coating the pathogen, antibodies stimulate effector functions against the pathogen in cells that recognize their Fc region.[5]
Those cells that recognize coated pathogens have Fc receptors, which, as the name suggests, interact with the Fc region of IgA, IgG, and IgE antibodies. The engagement of a particular antibody with the Fc receptor on a particular cell triggers an effector function of that cell; phagocytes will phagocytose, mast cells and neutrophils will degranulate, natural killer cells will release cytokines and cytotoxic molecules; that will ultimately result in destruction of the invading microbe. The activation of natural killer cells by antibodies initiates a cytotoxic mechanism known as antibody-dependent cell-mediated cytotoxicity (ADCC) – this process may explain the efficacy of monoclonal antibodies used in biological therapies against cancer. The Fc receptors are isotype-specific, which gives greater flexibility to the immune system, invoking only the appropriate immune mechanisms for distinct pathogens.[2]
Natural antibodies
Humans and higher primates also produce "natural antibodies" that are present in serum before viral infection. Natural antibodies have been defined as antibodies that are produced without any previous infection, vaccination, other foreign antigen exposure or passive immunization. These antibodies can activate the classical complement pathway leading to lysis of enveloped virus particles long before the adaptive immune response is activated. Many natural antibodies are directed against the disaccharide galactose α(1,3)-galactose (α-Gal), which is found as a terminal sugar on glycosylated cell surface proteins, and generated in response to production of this sugar by bacteria contained in the human gut.[51] Rejection of xenotransplantated organs is thought to be, in part, the result of natural antibodies circulating in the serum of the recipient binding to α-Gal antigens expressed on the donor tissue.[52]
Immunoglobulin diversity
Virtually all microbes can trigger an antibody response. Successful recognition and eradication of many different types of microbes requires diversity among antibodies; their amino acid composition varies allowing them to interact with many different antigens.[53] It has been estimated that humans generate about 10 billion different antibodies, each capable of binding a distinct epitope of an antigen.[54] Although a huge repertoire of different antibodies is generated in a single individual, the number of genes available to make these proteins is limited by the size of the human genome. Several complex genetic mechanisms have evolved that allow vertebrate B cells to generate a diverse pool of antibodies from a relatively small number of antibody genes.[55]
Domain variability
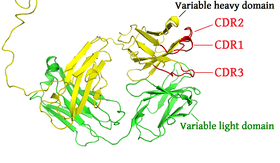
The chromosomal region that encodes an antibody is large and contains several distinct gene loci for each domain of the antibody—the chromosome region containing heavy chain genes (IGH@) is found on chromosome 14, and the loci containing lambda and kappa light chain genes (IGL@ and IGK@) are found on chromosomes 22 and 2 in humans. One of these domains is called the variable domain, which is present in each heavy and light chain of every antibody, but can differ in different antibodies generated from distinct B cells. Differences, between the variable domains, are located on three loops known as hypervariable regions (HV-1, HV-2 and HV-3) or complementarity determining regions (CDR1, CDR2 and CDR3). CDRs are supported within the variable domains by conserved framework regions. The heavy chain locus contains about 65 different variable domain genes that all differ in their CDRs. Combining these genes with an array of genes for other domains of the antibody generates a large cavalry of antibodies with a high degree of variability. This combination is called V(D)J recombination discussed below.[56]
V(D)J recombination
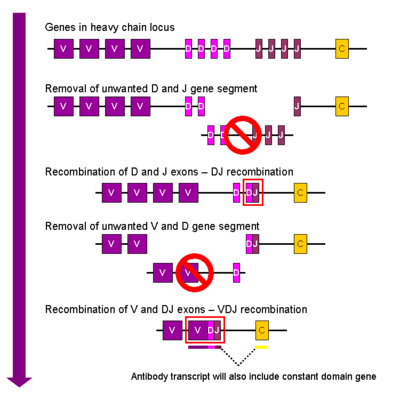
Somatic recombination of immunoglobulins, also known as V(D)J recombination, involves the generation of a unique immunoglobulin variable region. The variable region of each immunoglobulin heavy or light chain is encoded in several pieces—known as gene segments (subgenes). These segments are called variable (V), diversity (D) and joining (J) segments.[55] V, D and J segments are found in Ig heavy chains, but only V and J segments are found in Ig light chains. Multiple copies of the V, D and J gene segments exist, and are tandemly arranged in the genomes of mammals. In the bone marrow, each developing B cell will assemble an immunoglobulin variable region by randomly selecting and combining one V, one D and one J gene segment (or one V and one J segment in the light chain). As there are multiple copies of each type of gene segment, and different combinations of gene segments can be used to generate each immunoglobulin variable region, this process generates a huge number of antibodies, each with different paratopes, and thus different antigen specificities.[8] The rearrangement of several subgenes (i.e. V2 family) for lambda light chain immunoglobulin is coupled with the activation of microRNA miR-650, which further influences biology of B-cells.
RAG proteins play an important role with V(D)J recombination in cutting DNA at a particular region.[8] Without the presence of these proteins, V(D)J recombination would not occur.[8]
After a B cell produces a functional immunoglobulin gene during V(D)J recombination, it cannot express any other variable region (a process known as allelic exclusion) thus each B cell can produce antibodies containing only one kind of variable chain.[2][57]
Somatic hypermutation and affinity maturation
Following activation with antigen, B cells begin to proliferate rapidly. In these rapidly dividing cells, the genes encoding the variable domains of the heavy and light chains undergo a high rate of point mutation, by a process called somatic hypermutation (SHM). SHM results in approximately one nucleotide change per variable gene, per cell division.[10] As a consequence, any daughter B cells will acquire slight amino acid differences in the variable domains of their antibody chains.
This serves to increase the diversity of the antibody pool and impacts the antibody's antigen-binding affinity.[58] Some point mutations will result in the production of antibodies that have a weaker interaction (low affinity) with their antigen than the original antibody, and some mutations will generate antibodies with a stronger interaction (high affinity).[59] B cells that express high affinity antibodies on their surface will receive a strong survival signal during interactions with other cells, whereas those with low affinity antibodies will not, and will die by apoptosis.[59] Thus, B cells expressing antibodies with a higher affinity for the antigen will outcompete those with weaker affinities for function and survival allowing the average affinity of antibodies to increase over time. The process of generating antibodies with increased binding affinities is called affinity maturation. Affinity maturation occurs in mature B cells after V(D)J recombination, and is dependent on help from helper T cells.[60]
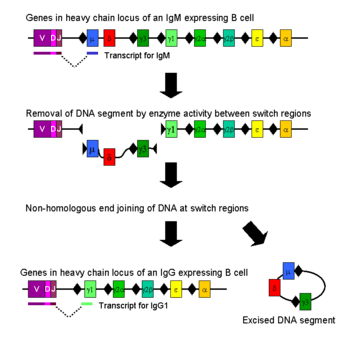
Class switching
Isotype or class switching is a biological process occurring after activation of the B cell, which allows the cell to produce different classes of antibody (IgA, IgE, or IgG).[8] The different classes of antibody, and thus effector functions, are defined by the constant (C) regions of the immunoglobulin heavy chain. Initially, naive B cells express only cell-surface IgM and IgD with identical antigen binding regions. Each isotype is adapted for a distinct function; therefore, after activation, an antibody with an IgG, IgA, or IgE effector function might be required to effectively eliminate an antigen. Class switching allows different daughter cells from the same activated B cell to produce antibodies of different isotypes. Only the constant region of the antibody heavy chain changes during class switching; the variable regions, and therefore antigen specificity, remain unchanged. Thus the progeny of a single B cell can produce antibodies, all specific for the same antigen, but with the ability to produce the effector function appropriate for each antigenic challenge. Class switching is triggered by cytokines; the isotype generated depends on which cytokines are present in the B cell environment.[61]
Class switching occurs in the heavy chain gene locus by a mechanism called class switch recombination (CSR). This mechanism relies on conserved nucleotide motifs, called switch (S) regions, found in DNA upstream of each constant region gene (except in the δ-chain). The DNA strand is broken by the activity of a series of enzymes at two selected S-regions.[62][63] The variable domain exon is rejoined through a process called non-homologous end joining (NHEJ) to the desired constant region (γ, α or ε). This process results in an immunoglobulin gene that encodes an antibody of a different isotype.[64]
Specificity designations
An antibody can be called monospecific if it has specificity for the same antigen or epitope,[65] or bispecific if they have affinity for two different antigens or two different epitopes on the same antigen.[66] A group of antibodies can be called polyvalent (or unspecific) if they have affinity for various antigens[67] or microorganisms.[67] Intravenous immunoglobulin, if not otherwise noted, consists of a variety of different IgG (polyclonal IgG). In contrast, monoclonal antibodies are identical antibodies produced by a single B cell.
Asymmetrical antibodies
Heterodimeric antibodies, which are also asymmetrical and antibodies, allow for greater flexibility and new formats for attaching a variety of drugs to the antibody arms. One of the general formats for a heterodimeric antibody is the “knobs-into-holes” format. This format is specific to the heavy chain part of the constant region in antibodies. The “knobs” part is engineered by replacing a small amino acid with a larger one. It fits into the “hole”, which is engineered by replacing a large amino acid with a smaller one. What connects the “knobs” to the “holes” are the disulfide bonds between each chain. The “knobs-into-holes” shape facilitates antibody dependent cell mediated cytotoxicity. Single chain variable fragments (scFv) are connected to the variable domain of the heavy and light chain via a short linker peptide. The linker is rich in glycine, which gives it more flexibility, and serine/threonine, which gives it specificity. Two different scFv fragments can be connected together, via a hinge region, to the constant domain of the heavy chain or the constant domain of the light chain.[68] This gives the antibody bispecificity, allowing for the binding specificities of two different antigens.[69] The “knobs-into-holes” format enhances heterodimer formation but doesn't suppress homodimer formation.
To further improve the function of heterodimeric antibodies, many scientists are looking towards artificial constructs. Artificial antibodies are largely diverse protein motifs that use the functional strategy of the antibody molecule, but aren't limited by the loop and framework structural constraints of the natural antibody.[70] Being able to control the combinational design of the sequence and three-dimensional space could transcend the natural design and allow for the attachment of different combinations of drugs to the arms.
Heterodimeric antibodies have a greater range in shapes they can take and the drugs that are attached to the arms don't have to be the same on each arm, allowing for different combinations of drugs to be used in cancer treatment. Pharmaceuticals are able to produce highly functional bispecific, and even multispecific, antibodies. The degree to which they can function is impressive given that such a change of shape from the natural form should lead to decreased functionality.
Medical applications
Disease diagnosis
Detection of particular antibodies is a very common form of medical diagnostics, and applications such as serology depend on these methods.[71] For example, in biochemical assays for disease diagnosis,[72] a titer of antibodies directed against Epstein-Barr virus or Lyme disease is estimated from the blood. If those antibodies are not present, either the person is not infected or the infection occurred a very long time ago, and the B cells generating these specific antibodies have naturally decayed.
In clinical immunology, levels of individual classes of immunoglobulins are measured by nephelometry (or turbidimetry) to characterize the antibody profile of patient.[73] Elevations in different classes of immunoglobulins are sometimes useful in determining the cause of liver damage in patients for whom the diagnosis is unclear.[1] For example, elevated IgA indicates alcoholic cirrhosis, elevated IgM indicates viral hepatitis and primary biliary cirrhosis, while IgG is elevated in viral hepatitis, autoimmune hepatitis and cirrhosis.
Autoimmune disorders can often be traced to antibodies that bind the body's own epitopes; many can be detected through blood tests. Antibodies directed against red blood cell surface antigens in immune mediated hemolytic anemia are detected with the Coombs test.[74] The Coombs test is also used for antibody screening in blood transfusion preparation and also for antibody screening in antenatal women.[74]
Practically, several immunodiagnostic methods based on detection of complex antigen-antibody are used to diagnose infectious diseases, for example ELISA, immunofluorescence, Western blot, immunodiffusion, immunoelectrophoresis, and magnetic immunoassay. Antibodies raised against human chorionic gonadotropin are used in over the counter pregnancy tests.
New dioxaborolane chemistry enables radioactive fluoride (18F) labeling of antibodies, which allows for positron emission tomography (PET) imaging of cancer.[75]
Disease therapy
Targeted monoclonal antibody therapy is employed to treat diseases such as rheumatoid arthritis,[76] multiple sclerosis,[77] psoriasis,[78] and many forms of cancer including non-Hodgkin's lymphoma,[79] colorectal cancer, head and neck cancer and breast cancer.[80]
Some immune deficiencies, such as X-linked agammaglobulinemia and hypogammaglobulinemia, result in partial or complete lack of antibodies.[81] These diseases are often treated by inducing a short term form of immunity called passive immunity. Passive immunity is achieved through the transfer of ready-made antibodies in the form of human or animal serum, pooled immunoglobulin or monoclonal antibodies, into the affected individual.[82]
Prenatal therapy
Rh factor, also known as Rh D antigen, is an antigen found on red blood cells; individuals that are Rh-positive (Rh+) have this antigen on their red blood cells and individuals that are Rh-negative (Rh–) do not. During normal childbirth, delivery trauma or complications during pregnancy, blood from a fetus can enter the mother's system. In the case of an Rh-incompatible mother and child, consequential blood mixing may sensitize an Rh- mother to the Rh antigen on the blood cells of the Rh+ child, putting the remainder of the pregnancy, and any subsequent pregnancies, at risk for hemolytic disease of the newborn.[83]
Rho(D) immune globulin antibodies are specific for human RhD antigen.[84] Anti-RhD antibodies are administered as part of a prenatal treatment regimen to prevent sensitization that may occur when a Rh-negative mother has a Rh-positive fetus. Treatment of a mother with Anti-RhD antibodies prior to and immediately after trauma and delivery destroys Rh antigen in the mother's system from the fetus. It is important to note that this occurs before the antigen can stimulate maternal B cells to "remember" Rh antigen by generating memory B cells. Therefore, her humoral immune system will not make anti-Rh antibodies, and will not attack the Rh antigens of the current or subsequent babies. Rho(D) Immune Globulin treatment prevents sensitization that can lead to Rh disease, but does not prevent or treat the underlying disease itself.[84]
Research applications
Specific antibodies are produced by injecting an antigen into a mammal, such as a mouse, rat, rabbit, goat, sheep, or horse for large quantities of antibody. Blood isolated from these animals contains polyclonal antibodies—multiple antibodies that bind to the same antigen—in the serum, which can now be called antiserum. Antigens are also injected into chickens for generation of polyclonal antibodies in egg yolk.[85] To obtain antibody that is specific for a single epitope of an antigen, antibody-secreting lymphocytes are isolated from the animal and immortalized by fusing them with a cancer cell line. The fused cells are called hybridomas, and will continually grow and secrete antibody in culture. Single hybridoma cells are isolated by dilution cloning to generate cell clones that all produce the same antibody; these antibodies are called monoclonal antibodies.[86] Polyclonal and monoclonal antibodies are often purified using Protein A/G or antigen-affinity chromatography.[87]
In research, purified antibodies are used in many applications. Antibodies for research applications can be found directly from antibody suppliers, or through use of a specialist search engine. Research antibodies are most commonly used to identify and locate intracellular and extracellular proteins. Antibodies are used in flow cytometry to differentiate cell types by the proteins they express; different types of cell express different combinations of cluster of differentiation molecules on their surface, and produce different intracellular and secretable proteins.[88] They are also used in immunoprecipitation to separate proteins and anything bound to them (co-immunoprecipitation) from other molecules in a cell lysate,[89] in Western blot analyses to identify proteins separated by electrophoresis,[90] and in immunohistochemistry or immunofluorescence to examine protein expression in tissue sections or to locate proteins within cells with the assistance of a microscope.[88][91] Proteins can also be detected and quantified with antibodies, using ELISA and ELISpot techniques.[92][93]
Antibodies used in research are some of the most powerful, yet most problematic reagents with a tremendous number of factors that must be controlled in any experiment including cross reactivity, or the antibody recognizing multiple epitopes and affinity, which can vary widely depending on experimental conditions such as pH, solvent, state of tissue etc. Multiple attempts have been made to improve both the way that researchers validate antibodies[94][95] and ways in which they report on antibodies. Researchers using antibodies in their work need to record them correctly in order to allow their research to be reproducible (and therefore tested, and qualified by other researchers). Less than half of research antibodies referenced in academic papers can be easily identified.[96] Papers published in F1000 in 2014 and 2015 provide researchers with a guide for reporting research antibody use.[97][98] The RRID paper, is co-published in 4 journals that implemented the RRIDs Standard for research resource citation, which draws data from the antibodyregistry.org as the source of antibody identifiers[99] (see also group at Force11[100]).
Regulations
Production and testing
Traditionally, most antibodies are produced by hybridoma cell lines through immortalization of antibody-producing cells by chemically-induced fusion with myeloma cells. In some cases, additional fusions with other lines have created "triomas" and "quadromas". The manufacturing process should be appropriately described and validated. Validation studies should at least include:
- The demonstration that the process is able to produce in good quality (the process should be validated)
- The efficiency of the antibody purification (all impurities and virus must be eliminated)
- The characterization of purified antibody (physicochemical characterization, immunological properties, biological activities, contaminants, ...)
- Determination of the virus clearance studies
Before clinical trials
- Product safety testing: Sterility (bacteria and fungi), In vitro and in vivo testing for adventitious viruses, Murine retrovirus testing... Product safety data needed before the initiation of feasibility trials in serious or immediately life-threatening conditions, it serves to evaluate dangerous potential of the product.
- Feasibility testing: These are pilot studies whose objectives include, among others, early characterization of safety and initial proof of concept in a small specific patient population (in vitro or in vivo testing).
Preclinical studies
- Testing cross-reactivity of antibody: to highlight unwanted interactions (toxicity) of antibodies with previously characterized tissues. This study can be performed in vitro (Reactivity of the antibody or immunoconjugate should be determined with a quick-frozen adult tissues) or in vivo (with appropriates animal models). More information about in vitro cross-reactivity testing.
- Preclinical pharmacology and toxicity testing: Preclinical safety testing of antibody is designed to identify possible toxicity in humans, to estimate the likelihood and severity of potential adverse events in humans, and to identify a safe starting dose and dose escalation, when possible.
- Animal toxicity studies: Acute toxicity testing, Repeat-dose toxicity testing, Long-term toxicity testing Safety & Testing | ari.info
- Pharmacokinetics and pharmacodynamics testing: Use for determinate clinical dosages, antibody activities (AUC, pharmacodynamics, biodistribution, ...), evaluation of the potential clinical effects
Structure prediction and computational antibody design
The importance of antibodies in health care and the biotechnology industry demands knowledge of their structures at high resolution. This information is used for protein engineering, modifying the antigen binding affinity, and identifying an epitope, of a given antibody. X-ray crystallography is one commonly used method for determining antibody structures. However, crystallizing an antibody is often laborious and time-consuming. Computational approaches provide a cheaper and faster alternative to crystallography, but their results are more equivocal, since they do not produce empirical structures. Online web servers such as Web Antibody Modeling (WAM)[101] and Prediction of Immunoglobulin Structure (PIGS)[102] enables computational modeling of antibody variable regions. Rosetta Antibody is a novel antibody FV region structure prediction server, which incorporates sophisticated techniques to minimize CDR loops and optimize the relative orientation of the light and heavy chains, as well as homology models that predict successful docking of antibodies with their unique antigen.[103]
The ability to describe the antibody through binding affinity to the antigen is supplemented by information on antibody structure and amino acid sequences for the purpose of patent claims.[104] Several methods have been presented for computational design of antibodies based on the structural bioinformatics studies of antibody CDRs.[105][106][107]
There are a variety of methods used to sequence an antibody including Edman degradation, cDNA, etc.; albeit one of the most common modern uses for peptide/protein identification is liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS).[108] High volume antibody sequencing methods require computational approaches for the data analysis, including de novo sequencing directly from tandem mass spectra[109] and database search methods that use existing protein sequence databases.[110][111] Many versions of shotgun protein sequencing are able to increase the coverage by utilizing CID/HCD/ETD[112] fragmentation methods and other techniques, and they have achieved substantial progress in attempt to fully sequence proteins, especially antibodies. Other methods have assumed the existence of similar proteins,[113] a known genome sequence,[114] or combined top-down and bottom up approaches.[115] Current technologies have the ability to assemble protein sequences with high accuracy by integrating de novo sequencing peptides, intensity, and positional confidence scores from database and homology searches.[116]
Antibody mimetic
Antibody mimetics are organic compounds, like antibodies, that can specifically bind antigens. They are usually artificial peptides or proteins with a molar mass of about 3 to 20 kDa. Nucleic acids and small molecules are sometimes considered antibody mimetics, but not artificial antibodies, antibody fragments and fusion proteins are composed from these. Common advantages over antibodies are better solubility, tissue penetration, stability towards heat and enzymes, and comparatively low production costs. Antibody mimetics such as the Affimer and the DARPin have being developed and commercialised as research, diagnostic and therapeutic agents.[117]
See also
- Affimer
- Antibody mimetic
- Anti-mitochondrial antibodies
- Anti-nuclear antibodies
- Aptamer
- Colostrum
- ELISA
- Humoral immunity
- Immunology
- Immunosuppressive drug
- Intravenous immunoglobulin (IVIg)
- Magnetic immunoassay
- Microantibody
- Monoclonal antibody
- Neutralizing antibody
- Secondary antibodies
- Single-domain antibody
- Slope spectroscopy
- Western blot normalization
References
- Rhoades RA, Pflanzer RG (2002). Human Physiology (5th ed.). Thomson Learning. p. 584. ISBN 978-0-534-42174-8.
- Janeway C (2001). Immunobiology (5th ed.). Garland Publishing. ISBN 978-0-8153-3642-6.
- Litman GW, Rast JP, Shamblott MJ, Haire RN, Hulst M, Roess W, Litman RT, Hinds-Frey KR, Zilch A, Amemiya CT (January 1993). "Phylogenetic diversification of immunoglobulin genes and the antibody repertoire". Molecular Biology and Evolution. 10 (1): 60–72. doi:10.1093/oxfordjournals.molbev.a040000. PMID 8450761.
- Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB (February 2015). "Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review". Journal of Autoimmunity. 57 (6): 1–13. doi:10.1016/j.jaut.2014.12.002. PMC 4340844. PMID 25578468.
- Pier GB, Lyczak JB, Wetzler LM (2004). Immunology, Infection, and Immunity. ASM Press. ISBN 978-1-55581-246-1.
- Borghesi L, Milcarek C (2006). "From B cell to plasma cell: regulation of V(D)J recombination and antibody secretion". Immunologic Research. 36 (1–3): 27–32. doi:10.1385/IR:36:1:27. PMID 17337763.
- Parker DC (1993). "T cell-dependent B cell activation". Annual Review of Immunology. 11 (1): 331–60. doi:10.1146/annurev.iy.11.040193.001555. PMID 8476565.
- Market E, Papavasiliou FN (October 2003). "V(D)J recombination and the evolution of the adaptive immune system". PLoS Biology. 1 (1): E16. doi:10.1371/journal.pbio.0000016. PMC 212695. PMID 14551913.
- Williams CM, Galli SJ (May 2000). "The diverse potential effector and immunoregulatory roles of mast cells in allergic disease". The Journal of Allergy and Clinical Immunology. 105 (5): 847–59. doi:10.1067/mai.2000.106485. PMID 10808163.
- Diaz M, Casali P (April 2002). "Somatic immunoglobulin hypermutation". Current Opinion in Immunology. 14 (2): 235–40. doi:10.1016/S0952-7915(02)00327-8. PMC 4621002. PMID 11869898.
- Lindenmann J (April 1984). "Origin of the terms 'antibody' and 'antigen'". Scandinavian Journal of Immunology. 19 (4): 281–5. doi:10.1111/j.1365-3083.1984.tb00931.x. PMID 6374880.
- Padlan EA (February 1994). "Anatomy of the antibody molecule". Molecular Immunology. 31 (3): 169–217. doi:10.1016/0161-5890(94)90001-9. PMID 8114766.
- "New Sculpture Portraying Human Antibody as Protective Angel Installed on Scripps Florida Campus". Archived from the original on 10 January 2011. Retrieved 12 December 2008.
- Pescovitz, David (22 October 2008). "Protein sculpture inspired by Vitruvian Man". Archived from the original on 4 November 2010. Retrieved 12 December 2008.
- "Emil von Behring — Biography". Archived from the original on 15 November 2010. Retrieved 5 June 2007.
- AGN (August 1931). "The Late Baron Shibasaburo Kitasato". Canadian Medical Association Journal. 25 (2): 206. PMC 382621. PMID 20318414.
- Winau F, Westphal O, Winau R (July 2004). "Paul Ehrlich--in search of the magic bullet". Microbes and Infection. 6 (8): 786–9. doi:10.1016/j.micinf.2004.04.003. PMID 15207826.
- Silverstein AM (May 2003). "Cellular versus humoral immunology: a century-long dispute". Nature Immunology. 4 (5): 425–8. doi:10.1038/ni0503-425. PMID 12719732.
- Van Epps HL (January 2006). "Michael Heidelberger and the demystification of antibodies" (PDF). The Journal of Experimental Medicine. 203 (1): 5. doi:10.1084/jem.2031fta. PMC 2118068. PMID 16523537. Archived (PDF) from the original on 9 August 2008.
- Marrack JR (1938). Chemistry of antigens and antibodies (2nd ed.). London: His Majesty's Stationery Office. OCLC 3220539.
- "The Linus Pauling Papers: How Antibodies and Enzymes Work". Archived from the original on 5 December 2010. Retrieved 5 June 2007.
- Silverstein AM (December 2004). "Labeled antigens and antibodies: the evolution of magic markers and magic bullets" (PDF). Nature Immunology. 5 (12): 1211–7. doi:10.1038/ni1140. PMID 15549122. Archived from the original (PDF) on 25 March 2009.
- Edelman GM, Gally JA (August 1962). "The nature of Bence-Jones proteins. Chemical similarities to polypetide chains of myeloma globulins and normal gamma-globulins". The Journal of Experimental Medicine. 116 (2): 207–27. doi:10.1084/jem.116.2.207. PMC 2137388. PMID 13889153.
- Stevens FJ, Solomon A, Schiffer M (July 1991). "Bence Jones proteins: a powerful tool for the fundamental study of protein chemistry and pathophysiology". Biochemistry. 30 (28): 6803–5. doi:10.1021/bi00242a001. PMID 2069946.
- Raju TN (September 1999). "The Nobel chronicles. 1972: Gerald M Edelman (b 1929) and Rodney R Porter (1917-85)". Lancet. 354 (9183): 1040. doi:10.1016/S0140-6736(05)76658-7. PMID 10501404.
- Hochman J, Inbar D, Givol D (March 1973). "An active antibody fragment (Fv) composed of the variable portions of heavy and light chains". Biochemistry. 12 (6): 1130–5. doi:10.1021/bi00730a018. PMID 4569769.
- Tomasi TB (October 1992). "The discovery of secretory IgA and the mucosal immune system". Immunology Today. 13 (10): 416–8. doi:10.1016/0167-5699(92)90093-M. PMID 1343085.
- Preud'homme JL, Petit I, Barra A, Morel F, Lecron JC, Lelièvre E (October 2000). "Structural and functional properties of membrane and secreted IgD". Molecular Immunology. 37 (15): 871–87. doi:10.1016/S0161-5890(01)00006-2. PMID 11282392.
- Johansson SG (2006). "The discovery of immunoglobulin E". Allergy and Asthma Proceedings. 27 (2 Suppl 1): S3–6. PMID 16722325.
- Hozumi N, Tonegawa S (October 1976). "Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions". Proceedings of the National Academy of Sciences of the United States of America. 73 (10): 3628–32. Bibcode:1976PNAS...73.3628H. doi:10.1073/pnas.73.10.3628. PMC 431171. PMID 824647.
- Maxwell Myer W (2004). Greer JG, Foerster J, Lukens JN, Rodgers GM, Paraskevas F (eds.). Wintrobe's clinical hematology (11 ed.). Hagerstown, MD: Lippincott Williams & Wilkins. pp. 453–456. ISBN 978-0-7817-3650-3.
- Tolar P, Sohn HW, Pierce SK (February 2008). "Viewing the antigen-induced initiation of B-cell activation in living cells". Immunological Reviews. 221 (1): 64–76. doi:10.1111/j.1600-065X.2008.00583.x. PMID 18275475.
- Woof JM, Burton DR (February 2004). "Human antibody-Fc receptor interactions illuminated by crystal structures". Nature Reviews. Immunology. 4 (2): 89–99. doi:10.1038/nri1266. PMID 15040582.
- Underdown BJ, Schiff JM (1986). "Immunoglobulin A: strategic defense initiative at the mucosal surface". Annual Review of Immunology. 4 (1): 389–417. doi:10.1146/annurev.iy.04.040186.002133. PMID 3518747.
- Geisberger R, Lamers M, Achatz G (August 2006). "The riddle of the dual expression of IgM and IgD". Immunology. 118 (4): 429–37. doi:10.1111/j.1365-2567.2006.02386.x. PMC 1782314. PMID 16895553.
- Chen K, Xu W, Wilson M, He B, Miller NW, Bengtén E, Edholm ES, Santini PA, Rath P, Chiu A, Cattalini M, Litzman J, B Bussel J, Huang B, Meini A, Riesbeck K, Cunningham-Rundles C, Plebani A, Cerutti A (August 2009). "Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils". Nature Immunology. 10 (8): 889–98. doi:10.1038/ni.1748. PMC 2785232. PMID 19561614.
- Goding JW (1978). Allotypes of IgM and IgD receptors in the mouse: a probe for lymphocyte differentiation. Contemporary Topics in Immunobiology. 8. pp. 203–43. doi:10.1007/978-1-4684-0922-2_7. ISBN 978-1-4684-0924-6. PMID 357078.
- Lundqvist ML, Middleton DL, Radford C, Warr GW, Magor KE (2006). "Immunoglobulins of the non-galliform birds: antibody expression and repertoire in the duck". Developmental and Comparative Immunology. 30 (1–2): 93–100. doi:10.1016/j.dci.2005.06.019. PMC 1317265. PMID 16150486.
- Berstein RM, Schluter SF, Shen S, Marchalonis JJ (April 1996). "A new high molecular weight immunoglobulin class from the carcharhine shark: implications for the properties of the primordial immunoglobulin". Proceedings of the National Academy of Sciences of the United States of America. 93 (8): 3289–93. Bibcode:1996PNAS...93.3289B. doi:10.1073/pnas.93.8.3289. PMC 39599. PMID 8622930.
- Reth M. "Matching cellular dimensions with molecular sizes" (PDF).
- Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, Rudd PM, Woof JM, Dwek RA (January 1998). "The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcα receptor interactions". The Journal of Biological Chemistry. 273 (4): 2260–72. doi:10.1074/jbc.273.4.2260. PMID 9442070.
- Roux KH (October 1999). "Immunoglobulin structure and function as revealed by electron microscopy". International Archives of Allergy and Immunology. 120 (2): 85–99. doi:10.1159/000024226. PMID 10545762.
- Barclay AN (August 2003). "Membrane proteins with immunoglobulin-like domains--a master superfamily of interaction molecules". Seminars in Immunology. 15 (4): 215–23. doi:10.1016/S1044-5323(03)00047-2. PMID 14690046.
- Putnam FW, Liu YS, Low TL (April 1979). "Primary structure of a human IgA1 immunoglobulin. IV. Streptococcal IgA1 protease, digestion, Fab and Fc fragments, and the complete amino acid sequence of the alpha 1 heavy chain". The Journal of Biological Chemistry. 254 (8): 2865–74. PMID 107164.
- Al-Lazikani B, Lesk AM, Chothia C (November 1997). "Standard conformations for the canonical structures of immunoglobulins". Journal of Molecular Biology. 273 (4): 927–48. doi:10.1006/jmbi.1997.1354. PMID 9367782.
- North B, Lehmann A, Dunbrack RL (February 2011). "A new clustering of antibody CDR loop conformations". Journal of Molecular Biology. 406 (2): 228–56. doi:10.1016/j.jmb.2010.10.030. PMC 3065967. PMID 21035459.
- Nikoloudis D, Pitts JE, Saldanha JW (2014). "A complete, multi-level conformational clustering of antibody complementarity-determining regions". PeerJ. 2 (e456): e456. doi:10.7717/peerj.456. PMC 4103072. PMID 25071986.
- Heyman B (December 1996). "Complement and Fc-receptors in regulation of the antibody response". Immunology Letters. 54 (2–3): 195–9. doi:10.1016/S0165-2478(96)02672-7. PMID 9052877.
- Ravetch JV, Bolland S (2001). "IgG Fc receptors". Annual Review of Immunology. 19 (1): 275–90. doi:10.1146/annurev.immunol.19.1.275. PMID 11244038.
- Rus H, Cudrici C, Niculescu F (2005). "The role of the complement system in innate immunity". Immunologic Research. 33 (2): 103–12. doi:10.1385/IR:33:2:103. PMID 16234578.
- Racaniello, Vincent (6 October 2009). "Natural antibody protects against viral infection". Virology Blog. Archived from the original on 20 February 2010. Retrieved 22 January 2010.
- Milland J, Sandrin MS (December 2006). "ABO blood group and related antigens, natural antibodies and transplantation". Tissue Antigens. 68 (6): 459–66. doi:10.1111/j.1399-0039.2006.00721.x. PMID 17176435.
- Mian IS, Bradwell AR, Olson AJ (January 1991). "Structure, function and properties of antibody binding sites". Journal of Molecular Biology. 217 (1): 133–51. doi:10.1016/0022-2836(91)90617-F. PMID 1988675.
- Fanning LJ, Connor AM, Wu GE (April 1996). "Development of the immunoglobulin repertoire". Clinical Immunology and Immunopathology. 79 (1): 1–14. doi:10.1006/clin.1996.0044. PMID 8612345.
- Nemazee D (October 2006). "Receptor editing in lymphocyte development and central tolerance". Nature Reviews. Immunology. 6 (10): 728–40. doi:10.1038/nri1939. PMID 16998507.
- Peter Parham. "The Immune System. 2nd ed. Garland Science: New York, 2005. pg.47–62
- Bergman Y, Cedar H (October 2004). "A stepwise epigenetic process controls immunoglobulin allelic exclusion". Nature Reviews. Immunology. 4 (10): 753–61. doi:10.1038/nri1458. PMID 15459667.
- Honjo T, Habu S (1985). "Origin of immune diversity: genetic variation and selection". Annual Review of Biochemistry. 54 (1): 803–30. doi:10.1146/annurev.bi.54.070185.004103. PMID 3927822.
- Or-Guil M, Wittenbrink N, Weiser AA, Schuchhardt J (April 2007). "Recirculation of germinal center B cells: a multilevel selection strategy for antibody maturation". Immunological Reviews. 216: 130–41. doi:10.1111/j.1600-065X.2007.00507.x. PMID 17367339.
- Neuberger MS, Ehrenstein MR, Rada C, Sale J, Batista FD, Williams G, Milstein C (March 2000). "Memory in the B-cell compartment: antibody affinity maturation". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 355 (1395): 357–60. doi:10.1098/rstb.2000.0573. PMC 1692737. PMID 10794054.
- Stavnezer J, Amemiya CT (August 2004). "Evolution of isotype switching". Seminars in Immunology. 16 (4): 257–75. doi:10.1016/j.smim.2004.08.005. PMID 15522624.
- Durandy A (August 2003). "Activation-induced cytidine deaminase: a dual role in class-switch recombination and somatic hypermutation". European Journal of Immunology. 33 (8): 2069–73. doi:10.1002/eji.200324133. PMID 12884279.
- Casali P, Zan H (November 2004). "Class switching and Myc translocation: how does DNA break?". Nature Immunology. 5 (11): 1101–3. doi:10.1038/ni1104-1101. PMC 4625794. PMID 15496946.
- Lieber MR, Yu K, Raghavan SC (September 2006). "Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations". DNA Repair. 5 (9–10): 1234–45. doi:10.1016/j.dnarep.2006.05.013. PMID 16793349.
- page 22 in: Shoenfeld Y, Meroni P, Gershwin ME (2007). Autoantibodie. Amsterdam; Boston: Elsevier. ISBN 978-0-444-52763-9.
- Spiess C, Zhai Q, Carter PJ (October 2015). "Alternative molecular formats and therapeutic applications for bispecific antibodies". Molecular Immunology. 67 (2 Pt A): 95–106. doi:10.1016/j.molimm.2015.01.003. PMID 25637431.
- Farlex dictionary > polyvalent Citing: The American Heritage Medical Dictionary. 2004
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, Sweet H, Foltz IN, Wittekind M, Yan W (June 2010). "Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG". The Journal of Biological Chemistry. 285 (25): 19637–46. doi:10.1074/jbc.M110.117382. PMC 2885242. PMID 20400508.
- Muller KM (1998). "The first constant domain (CH1 and CL) of an antibody used as heterodimerization domain for bispecific miniantibodies". FEBS Letters. 422 (2): 259–264. doi:10.1016/s0014-5793(98)00021-0. PMID 9490020.
- Gao C, Mao S, Lo CH, Wirsching P, Lerner RA, Janda KD (May 1999). "Making artificial antibodies: a format for phage display of combinatorial heterodimeric arrays". Proceedings of the National Academy of Sciences of the United States of America. 96 (11): 6025–30. Bibcode:1999PNAS...96.6025G. doi:10.1073/pnas.96.11.6025. PMC 26829. PMID 10339535.
- "Animated depictions of how antibodies are used in ELISA assays". Cellular Technology Ltd.—Europe. Archived from the original on 14 June 2011. Retrieved 8 May 2007.
- "Animated depictions of how antibodies are used in ELISPOT assays". Cellular Technology Ltd.—Europe. Archived from the original on 16 May 2011. Retrieved 8 May 2007.
- Stern P (2006). "Current possibilities of turbidimetry and nephelometry" (PDF). Klin Biochem Metab. 14 (3): 146–151. Archived from the original (PDF) on 10 April 2008.
- Dean L (2005). "Chapter 4: Hemolytic disease of the newborn". Blood Groups and Red Cell Antigens. NCBI Bethesda (MD): National Library of Medicine (US).
- Rodriguez EA, Wang Y, Crisp JL, Vera DR, Tsien RY, Ting R (May 2016). "New Dioxaborolane Chemistry Enables [(18)F]-Positron-Emitting, Fluorescent [(18)F]-Multimodality Biomolecule Generation from the Solid Phase". Bioconjugate Chemistry. 27 (5): 1390–1399. doi:10.1021/acs.bioconjchem.6b00164. PMC 4916912. PMID 27064381.
- Feldmann M, Maini RN (2001). "Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned?". Annual Review of Immunology. 19 (1): 163–96. doi:10.1146/annurev.immunol.19.1.163. PMID 11244034.
- Doggrell SA (June 2003). "Is natalizumab a breakthrough in the treatment of multiple sclerosis?". Expert Opinion on Pharmacotherapy. 4 (6): 999–1001. doi:10.1517/14656566.4.6.999. PMID 12783595.
- Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, Dooley LT, Lebwohl M (February 2007). "A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis". The New England Journal of Medicine. 356 (6): 580–92. doi:10.1056/NEJMoa062382. PMID 17287478.
- Plosker GL, Figgitt DP (2003). "Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia". Drugs. 63 (8): 803–43. doi:10.2165/00003495-200363080-00005. PMID 12662126.
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ (2001). "First-line Herceptin monotherapy in metastatic breast cancer". Oncology. 61. 61 Suppl 2 (Suppl. 2): 37–42. doi:10.1159/000055400. PMID 11694786.
- LeBien TW (July 2000). "Fates of human B-cell precursors". Blood. 96 (1): 9–23. PMID 10891425. Archived from the original on 29 April 2010.
- Ghaffer A (26 March 2006). "Immunization". Immunology — Chapter 14. University of South Carolina School of Medicine. Archived from the original on 18 October 2010. Retrieved 6 June 2007.
- Urbaniak SJ, Greiss MA (March 2000). "RhD haemolytic disease of the fetus and the newborn". Blood Reviews. 14 (1): 44–61. doi:10.1054/blre.1999.0123. PMID 10805260.
- Fung Kee Fung K, Eason E, Crane J, Armson A, De La Ronde S, Farine D, Keenan-Lindsay L, Leduc L, Reid GJ, Aerde JV, Wilson RD, Davies G, Désilets VA, Summers A, Wyatt P, Young DC (September 2003). "Prevention of Rh alloimmunization". Journal of Obstetrics and Gynaecology Canada. 25 (9): 765–73. doi:10.1016/S1701-2163(16)31006-4. PMID 12970812.
- Tini M, Jewell UR, Camenisch G, Chilov D, Gassmann M (March 2002). "Generation and application of chicken egg-yolk antibodies". Comparative Biochemistry and Physiology. Part A, Molecular & Integrative Physiology. 131 (3): 569–74. doi:10.1016/S1095-6433(01)00508-6. PMID 11867282.
- Cole SP, Campling BG, Atlaw T, Kozbor D, Roder JC (June 1984). "Human monoclonal antibodies". Molecular and Cellular Biochemistry. 62 (2): 109–20. doi:10.1007/BF00223301. PMID 6087121.
- Kabir S (2002). "Immunoglobulin purification by affinity chromatography using protein A mimetic ligands prepared by combinatorial chemical synthesis". Immunological Investigations. 31 (3–4): 263–78. doi:10.1081/IMM-120016245. PMID 12472184.
- Brehm-Stecher BF, Johnson EA (September 2004). "Single-cell microbiology: tools, technologies, and applications". Microbiology and Molecular Biology Reviews. 68 (3): 538–59, table of contents. doi:10.1128/MMBR.68.3.538-559.2004. PMC 515252. PMID 15353569.
- Williams NE (2000). Immunoprecipitation procedures. Methods in Cell Biology. 62. San Diego, CA : Academic Press. pp. 449–53. doi:10.1016/S0091-679X(08)61549-6. ISBN 978-0-12-544164-3. PMID 10503210.
- Kurien BT, Scofield RH (April 2006). "Western blotting". Methods. 38 (4): 283–93. doi:10.1016/j.ymeth.2005.11.007. PMID 16483794.
- Scanziani E (1998). Immunohistochemical staining of fixed tissues. Methods in Molecular Biology. 104. Totowa, N.J. : Humana Press. pp. 133–40. doi:10.1385/0-89603-525-5:133. ISBN 978-0-89603-525-6. PMID 9711649.
- Reen DJ (1994). Enzyme-linked immunosorbent assay (ELISA). Methods in Molecular Biology. 32. pp. 461–6. doi:10.1385/0-89603-268-X:461. ISBN 978-0-89603-268-2. PMC 2366430. PMID 7951745.
- Kalyuzhny AE (2005). Chemistry and biology of the ELISPOT assay. Methods in Molecular Biology. 302. pp. 15–31. doi:10.1385/1-59259-903-6:015. ISBN 978-1-59259-903-5. PMID 15937343.
- Saper CB (December 2005). "An open letter to our readers on the use of antibodies". The Journal of Comparative Neurology. 493 (4): 477–8. doi:10.1002/cne.20839. PMID 16304632.
- "NOT-OD-16-011: Implementing Rigor and Transparency in NIH & AHRQ Research Grant Applications". grants.nih.gov.
- Vasilevsky, Nicole A.; Brush, Matthew H.; Paddock, Holly; Ponting, Laura; Tripathy, Shreejoy J.; Larocca, Gregory M.; Haendel, Melissa A. (2 September 2013). "On the reproducibility of science: unique identification of research resources in the biomedical literature". PeerJ. 1: e148. doi:10.7717/peerj.148. PMC 3771067. PMID 24032093.
- Bandrowski A, Brush M, Grethe JS, Haendel MA, Kennedy DN, Hill S, Hof PR, Martone ME, Pols M, Tan S, Washington N, Zudilova-Seinstra E, Vasilevsky N (2015). "The Resource Identification Initiative: A cultural shift in publishing". F1000Research. 4: 134. doi:10.12688/f1000research.6555.2. PMC 4648211. PMID 26594330.
- Helsby, Matthew A.; Fenn, Joe R.; Chalmers, Andrew D. (23 August 2013). "Reporting research antibody use: how to increase experimental reproducibility". F1000Research. 2: 153. doi:10.12688/f1000research.2-153.v2. PMC 3829129. PMID 24358895.
- "The Antibody Registry". antibodyregistry.org.
- "Resource Identification Initiative". FORCE11. 14 August 2013. Retrieved 18 April 2016.
- Archived 17 July 2011 at the Wayback Machine
WAM - Marcatili P, Rosi A, Tramontano A (September 2008). "PIGS: automatic prediction of antibody structures". Bioinformatics. 24 (17): 1953–4. doi:10.1093/bioinformatics/btn341. PMID 18641403. Archived from the original on 26 November 2010.
Prediction of Immunoglobulin Structure (PIGS) - Archived 19 July 2011 at the Wayback Machine
RosettaAntibody - Park, Hyeongsu. "Written Description Problems of the Monoclonal Antibody Patents after Centocor v. Abbott". jolt.law.harvard.edu. Archived from the original on 13 December 2014. Retrieved 12 December 2014.
- Adolf-Bryfogle, J; Kalyuzhniy, O; Kubitz, M; Weitzner, BD; Hu, X; Adachi, Y; Schief, WR; Dunbrack, RL (April 2018). "RosettaAntibodyDesign (RAbD): A general framework for computational antibody design". PLOS Computational Biology. 14 (4): e1006112. Bibcode:2018PLSCB..14E6112A. doi:10.1371/journal.pcbi.1006112. PMC 5942852. PMID 29702641.
- Lapidoth, GD; Baran, D; Pszolla, GM; Norn, C; Alon, A; Tyka, MD; Fleishman, SJ (August 2015). "AbDesign: An algorithm for combinatorial backbone design guided by natural conformations and sequences". Proteins. 83 (8): 1385–406. doi:10.1002/prot.24779. PMC 4881815. PMID 25670500.
- Li, T; Pantazes, RJ; Maranas, CD (2014). "OptMAVEn--a new framework for the de novo design of antibody variable region models targeting specific antigen epitopes". PLOS ONE. 9 (8): e105954. Bibcode:2014PLoSO...9j5954L. doi:10.1371/journal.pone.0105954. PMC 4143332. PMID 25153121.
- Pham, Victoria; Henzel, William J.; Arnott, David; Hymowitz, Sarah; Sandoval, Wendy N.; Truong, Bao-Tran; Lowman, Henry; Lill, Jennie R. (2006). "De novo proteomic sequencing of a monoclonal antibody raised against OX40 ligand". Analytical Biochemistry. 352 (1): 77–86. doi:10.1016/j.ab.2006.02.001. PMID 16545334.
- Ma, Bin; Zhang, Kaizhong; Hendrie, Christopher; Liang, Chengzhi; Li, Ming; Doherty-Kirby, Amanda; Lajoie, Gilles (2003). "PEAKS: Powerful software for peptidede novo sequencing by tandem mass spectrometry". Rapid Communications in Mass Spectrometry. 17 (20): 2337–2342. Bibcode:2003RCMS...17.2337M. doi:10.1002/rcm.1196. PMID 14558135.
- Zhang, Jing; Xin, Lei; Shan, Baozhen; Chen, Weiwu; Xie, Mingjie; Yuen, Denis; Zhang, Weiming; Zhang, Zefeng; Lajoie, Gilles A.; Ma, Bin (2012). "PEAKS DB:De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification". Molecular & Cellular Proteomics. 11 (4): M111.010587. doi:10.1074/mcp.M111.010587. PMID 22186715.
- Perkins, David N.; Pappin, Darryl J. C.; Creasy, David M.; Cottrell, John S. (1999). "Probability-based protein identification by searching sequence databases using mass spectrometry data". Electrophoresis. 20 (18): 3551–3567. doi:10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2.
- Bandeira, Nuno; Tang, Haixu; Bafna, Vineet; Pevzner, Pavel (2004). "Shotgun Protein Sequencing by Tandem Mass Spectra Assembly". Analytical Chemistry. 76 (24): 7221–7233. doi:10.1021/ac0489162. PMID 15595863.
- Liu, Xiaowen; Han, Yonghua; Yuen, Denis; Ma, Bin (2009). "Automated protein (Re)sequencing with MS/MS and a homologous database yields almost full coverage and accuracy". Bioinformatics. 25 (17): 2174–2180. doi:10.1093/bioinformatics/btp366. PMID 19535534.
- Castellana, Natalie E.; Pham, Victoria; Arnott, David; Lill, Jennie R.; Bafna, Vineet (2010). "Template Proteogenomics: Sequencing Whole Proteins Using an Imperfect Database". Molecular & Cellular Proteomics. 9 (6): 1260–1270. doi:10.1074/mcp.M900504-MCP200. PMID 20164058.
- Liu, Xiaowen; Dekker, Lennard J. M.; Wu, Si; Vanduijn, Martijn M.; Luider, Theo M.; Tolić, Nikola; Kou, Qiang; Dvorkin, Mikhail; Alexandrova, Sonya; Vyatkina, Kira; Paša-Tolić, Ljiljana; Pevzner, Pavel A. (2014). "De Novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra". Journal of Proteome Research. 13 (7): 3241–3248. doi:10.1021/pr401300m. PMID 24874765.
- Tran, Ngoc Hieu; Rahman, M. Ziaur; He, Lin; Xin, Lei; Shan, Baozhen; Li, Ming (2016). "Complete de Novo Assembly of Monoclonal Antibody Sequences". Scientific Reports. 6: 31730. Bibcode:2016NatSR...631730T. doi:10.1038/srep31730. PMID 27562653.
- Gebauer M, Skerra A (June 2009). "Engineered protein scaffolds as next-generation antibody therapeutics". Current Opinion in Chemical Biology. 13 (3): 245–55. doi:10.1016/j.cbpa.2009.04.627. PMID 19501012.
External links
| Wikimedia Commons has media related to Antibodies. |
- Mike's Immunoglobulin Structure/Function Page at University of Cambridge
- Antibodies as the PDB molecule of the month Discussion of the structure of antibodies at RCSB Protein Data Bank
- Microbiology and Immunology On-line Textbook at University of South Carolina
- A hundred years of antibody therapy History and applications of antibodies in the treatment of disease at University of Oxford
- How Lymphocytes Produce Antibody from Cells Alive!
- Antibody applications Fluorescent antibody image library, University of Birmingham