Inflammation
Inflammation (from Latin: inflammatio) is part of the complex biological response of body tissues to harmful stimuli, such as pathogens, damaged cells, or irritants,[1] and is a protective response involving immune cells, blood vessels, and molecular mediators. The function of inflammation is to eliminate the initial cause of cell injury, clear out necrotic cells and tissues damaged from the original insult and the inflammatory process, and initiate tissue repair.
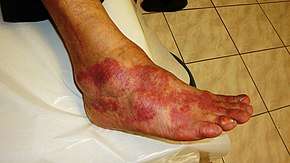
The five classical signs of inflammation are heat, pain, redness, swelling, and loss of function (Latin calor, dolor, rubor, tumor, and functio laesa). Inflammation is a generic response, and therefore it is considered as a mechanism of innate immunity, as compared to adaptive immunity, which is specific for each pathogen.[2] Too little inflammation could lead to progressive tissue destruction by the harmful stimulus (e.g. bacteria) and compromise the survival of the organism. In contrast, chronic inflammation is associated with various diseases, such as hay fever, periodontal disease, atherosclerosis, and osteoarthritis.
Inflammation can be classified as either acute or chronic. Acute inflammation is the initial response of the body to harmful stimuli and is achieved by the increased movement of plasma and leukocytes (especially granulocytes) from the blood into the injured tissues. A series of biochemical events propagates and matures the inflammatory response, involving the local vascular system, the immune system, and various cells within the injured tissue. Prolonged inflammation, known as chronic inflammation, leads to a progressive shift in the type of cells present at the site of inflammation, such as mononuclear cells, and is characterized by simultaneous destruction and healing of the tissue from the inflammatory process.
Inflammation is not a synonym for infection. Infection describes the interaction between the action of microbial invasion and the reaction of the body's inflammatory response—the two components are considered together when discussing an infection, and the word is used to imply a microbial invasive cause for the observed inflammatory reaction. Inflammation on the other hand describes purely the body's immunovascular response, whatever the cause may be. But because of how often the two are correlated, words ending in the suffix -itis (which refers to inflammation) are sometimes informally described as referring to infection. For example, the word urethritis strictly means only "urethral inflammation", but clinical health care providers usually discuss urethritis as a urethral infection because urethral microbial invasion is the most common cause of urethritis.
It is useful to differentiate inflammation and infection because there are typical situations in pathology and medical diagnosis where inflammation is not driven by microbial invasion – for example, atherosclerosis, trauma, ischemia, and autoimmune diseases including type III hypersensitivity. Conversely, there is pathology where microbial invasion does not cause the classic inflammatory response – for example, parasitosis or eosinophilia.
Causes
- Burns[3]
- Frostbite
- Physical injury, blunt or penetrating[4]
- Foreign bodies, including splinters, dirt and debris
- Trauma[3]
- Ionizing radiation
Biological:
- Infection by pathogens[3]
- Immune reactions due to hypersensitivity
- Stress
Chemical:[3]
Psychological:
- Excitement[5]
Types
|
| Acute | Chronic | |
|---|---|---|
| Causative agent | Bacterial pathogens, injured tissues | Persistent acute inflammation due to non-degradable pathogens, viral infection, persistent foreign bodies, or autoimmune reactions |
| Major cells involved | neutrophils (primarily), basophils (inflammatory response), and eosinophils (response to helminth worms and parasites), mononuclear cells (monocytes, macrophages) | Mononuclear cells (monocytes, macrophages, lymphocytes, plasma cells), fibroblasts |
| Primary mediators | Vasoactive amines, eicosanoids | IFN-γ and other cytokines, growth factors, reactive oxygen species, hydrolytic enzymes |
| Onset | Immediate | Delayed |
| Duration | Few days | Up to many months, or years |
| Outcomes | Resolution, abscess formation, chronic inflammation | Tissue destruction, fibrosis, necrosis |
Cardinal signs
| English | Latin | |
|---|---|---|
| Redness | Rubor* | |
| Swelling | Tumor* | |
| Heat | Calor* | |
| Pain | Dolor* | |
| Loss of function | Functio laesa** | |
| All the above signs may be observed in specific instances, but no single sign must, as a matter of course, be present.[6]
These are the original, or "cardinal signs" of inflammation.[6]* Functio laesa is an antiquated notion, as it is not unique to inflammation and is a characteristic of many disease states.[7]** | ||
Acute inflammation is a short-term process, usually appearing within a few minutes or hours and begins to cease upon the removal of the injurious stimulus.[8] It involves a coordinated and systemic mobilization response locally of various immune, endocrine and neurological mediators of acute inflammation. In a normal healthy response, it becomes activated, clears the pathogen and begins a repair process and then ceases.[9] It is characterized by five cardinal signs:[10]
An acronym that may be used to remember the key symptoms is "PRISH", for pain, redness, immobility (loss of function), swelling and heat.
The traditional names for signs of inflammation come from Latin:
The first four (classical signs) were described by Celsus (ca. 30 BC–38 AD),[12] while loss of function was probably added later by Galen.[13] However, the addition of this fifth sign has also been ascribed to Thomas Sydenham[14] and Virchow.[8][10]
Redness and heat are due to increased blood flow at body core temperature to the inflamed site; swelling is caused by accumulation of fluid; pain is due to the release of chemicals such as bradykinin and histamine that stimulate nerve endings. Loss of function has multiple causes.[10]
Acute inflammation of the lung (usually caused in response to pneumonia) does not cause pain unless the inflammation involves the parietal pleura, which does have pain-sensitive nerve endings.[10]
Process of acute inflammation
The process of acute inflammation is initiated by resident immune cells already present in the involved tissue, mainly resident macrophages, dendritic cells, histiocytes, Kupffer cells and mast cells. These cells possess surface receptors known as pattern recognition receptors (PRRs), which recognize (i.e., bind) two subclasses of molecules: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). PAMPs are compounds that are associated with various pathogens, but which are distinguishable from host molecules. DAMPs are compounds that are associated with host-related injury and cell damage.
At the onset of an infection, burn, or other injuries, these cells undergo activation (one of the PRRs recognize a PAMP or DAMP) and release inflammatory mediators responsible for the clinical signs of inflammation. Vasodilation and its resulting increased blood flow causes the redness (rubor) and increased heat (calor). Increased permeability of the blood vessels results in an exudation (leakage) of plasma proteins and fluid into the tissue (edema), which manifests itself as swelling (tumor). Some of the released mediators such as bradykinin increase the sensitivity to pain (hyperalgesia, dolor). The mediator molecules also alter the blood vessels to permit the migration of leukocytes, mainly neutrophils and macrophages, outside of the blood vessels (extravasation) into the tissue. The neutrophils migrate along a chemotactic gradient created by the local cells to reach the site of injury.[8] The loss of function (functio laesa) is probably the result of a neurological reflex in response to pain.
In addition to cell-derived mediators, several acellular biochemical cascade systems consisting of preformed plasma proteins act in parallel to initiate and propagate the inflammatory response. These include the complement system activated by bacteria and the coagulation and fibrinolysis systems activated by necrosis, e.g. a burn or a trauma.[8]
Acute inflammation may be regarded as the first line of defense against injury. Acute inflammatory response requires constant stimulation to be sustained. Inflammatory mediators are short-lived and are quickly degraded in the tissue. Hence, acute inflammation begins to cease once the stimulus has been removed.[8]
Vascular component
Vasodilation and increased permeability
As defined, acute inflammation is an immunovascular response to an inflammatory stimulus. This means acute inflammation can be broadly divided into a vascular phase that occurs first, followed by a cellular phase involving immune cells (more specifically myeloid granulocytes in the acute setting). The vascular component of acute inflammation involves the movement of plasma fluid, containing important proteins such as fibrin and immunoglobulins (antibodies), into inflamed tissue.
Upon contact with PAMPs, tissue macrophages and mastocytes release vasoactive amines such as histamine and serotonin, as well as eicosanoids such as prostaglandin E2 and leukotriene B4 to remodel the local vasculature. Macrophages and endothelial cells release nitric oxide. These mediators vasodilate and permeabilize the blood vessels, which results in the net distribution of blood plasma from the vessel into the tissue space. The increased collection of fluid into the tissue causes it to swell (edema). This exuded tissue fluid contain various antimicrobial mediators from the plasma such as complement, lysozyme, antibodies, which can immediately deal damage to microbes, and opsonise the microbes in preparation for the cellular phase. If the inflammatory stimulus is a lacerating wound, exuded platelets, coagulants, plasmin and kinins can clot the wounded area and provide haemostasis in the first instance. These clotting mediators also provide a structural staging framework at the inflammatory tissue site in the form of a fibrin lattice – as would construction scaffolding at a construction site – for the purpose of aiding phagocytic debridement and wound repair later on. Some of the exuded tissue fluid is also funnelled by lymphatics to the regional lymph nodes, flushing bacteria along to start the recognition and attack phase of the adaptive immune system.
Acute inflammation is characterized by marked vascular changes, including vasodilation, increased permeability and increased blood flow, which are induced by the actions of various inflammatory mediators. Vasodilation occurs first at the arteriole level, progressing to the capillary level, and brings about a net increase in the amount of blood present, causing the redness and heat of inflammation. Increased permeability of the vessels results in the movement of plasma into the tissues, with resultant stasis due to the increase in the concentration of the cells within blood – a condition characterized by enlarged vessels packed with cells. Stasis allows leukocytes to marginate (move) along the endothelium, a process critical to their recruitment into the tissues. Normal flowing blood prevents this, as the shearing force along the periphery of the vessels moves cells in the blood into the middle of the vessel.
Plasma cascade systems
- The complement system, when activated, creates a cascade of chemical reactions that promotes opsonization, chemotaxis, and agglutination, and produces the MAC.
- The kinin system generates proteins capable of sustaining vasodilation and other physical inflammatory effects.
- The coagulation system or clotting cascade, which forms a protective protein mesh over sites of injury.
- The fibrinolysis system, which acts in opposition to the coagulation system, to counterbalance clotting and generate several other inflammatory mediators.
Plasma-derived mediators
* non-exhaustive list
| Name | Produced by | Description |
|---|---|---|
| Bradykinin | Kinin system | A vasoactive protein that is able to induce vasodilation, increase vascular permeability, cause smooth muscle contraction, and induce pain. |
| C3 | Complement system | Cleaves to produce C3a and C3b. C3a stimulates histamine release by mast cells, thereby producing vasodilation. C3b is able to bind to bacterial cell walls and act as an opsonin, which marks the invader as a target for phagocytosis. |
| C5a | Complement system | Stimulates histamine release by mast cells, thereby producing vasodilation. It is also able to act as a chemoattractant to direct cells via chemotaxis to the site of inflammation. |
| Factor XII (Hageman Factor) | Liver | A protein that circulates inactively, until activated by collagen, platelets, or exposed basement membranes via conformational change. When activated, it in turn is able to activate three plasma systems involved in inflammation: the kinin system, fibrinolysis system, and coagulation system. |
| Membrane attack complex | Complement system | A complex of the complement proteins C5b, C6, C7, C8, and multiple units of C9. The combination and activation of this range of complement proteins forms the membrane attack complex, which is able to insert into bacterial cell walls and causes cell lysis with ensuing bacterial death. |
| Plasmin | Fibrinolysis system | Able to break down fibrin clots, cleave complement protein C3, and activate Factor XII. |
| Thrombin | Coagulation system | Cleaves the soluble plasma protein fibrinogen to produce insoluble fibrin, which aggregates to form a blood clot. Thrombin can also bind to cells via the PAR1 receptor to trigger several other inflammatory responses, such as production of chemokines and nitric oxide. |
Cellular component
The cellular component involves leukocytes, which normally reside in blood and must move into the inflamed tissue via extravasation to aid in inflammation. Some act as phagocytes, ingesting bacteria, viruses, and cellular debris. Others release enzymatic granules that damage pathogenic invaders. Leukocytes also release inflammatory mediators that develop and maintain the inflammatory response. In general, acute inflammation is mediated by granulocytes, whereas chronic inflammation is mediated by mononuclear cells such as monocytes and lymphocytes.
Leukocyte extravasation

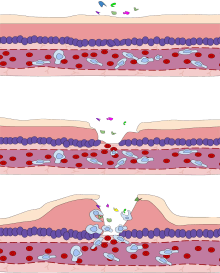
Various leukocytes, particularly neutrophils, are critically involved in the initiation and maintenance of inflammation. These cells must be able to move to the site of injury from their usual location in the blood, therefore mechanisms exist to recruit and direct leukocytes to the appropriate place. The process of leukocyte movement from the blood to the tissues through the blood vessels is known as extravasation, and can be broadly divided up into a number of steps:
- Leukocyte margination and endothelial adhesion: The white blood cells within the vessels which are generally centrally located move peripherally towards the walls of the vessels.[15] Activated macrophages in the tissue release cytokines such as IL-1 and TNFα, which in turn leads to production of chemokines that bind to proteoglycans forming gradient in the inflamed tissue and along the endothelial wall. Inflammatory cytokines induce the immediate expression of P-selectin on endothelial cell surfaces and P-selectin binds weakly to carbohydrate ligands on the surface of leukocytes and causes them to "roll" along the endothelial surface as bonds are made and broken. Cytokines released from injured cells induce the expression of E-selectin on endothelial cells, which functions similarly to P-selectin. Cytokines also induce the expression of integrin ligands such as ICAM-1 and VCAM-1 on endothelial cells, which mediate the adhesion and further slow leukocytes down. These weakly bound leukocytes are free to detach if not activated by chemokines produced in injured tissue after signal transduction via respective G protein-coupled receptors that activates integrins on the leukocyte surface for firm adhesion. Such activation increases the affinity of bound integrin receptors for ICAM-1 and VCAM-1 on the endothelial cell surface, firmly binding the leukocytes to the endothelium.
- Migration across the endothelium, known as transmigration, via the process of diapedesis: Chemokine gradients stimulate the adhered leukocytes to move between adjacent endothelial cells. The endothelial cells retract and the leukocytes pass through the basement membrane into the surrounding tissue using adhesion molecules such as ICAM-1.[15]
- Movement of leukocytes within the tissue via chemotaxis: Leukocytes reaching the tissue interstitium bind to extracellular matrix proteins via expressed integrins and CD44 to prevent them from leaving the site. A variety of molecules behave as chemoattractants, for example, C3a or C5, and cause the leukocytes to move along a chemotactic gradient towards the source of inflammation.
Phagocytosis
Extravasated neutrophils in the cellular phase come into contact with microbes at the inflamed tissue. Phagocytes express cell-surface endocytic pattern recognition receptors (PRRs) that have affinity and efficacy against non-specific microbe-associated molecular patterns (PAMPs). Most PAMPs that bind to endocytic PRRs and initiate phagocytosis are cell wall components, including complex carbohydrates such as mannans and β-glucans, lipopolysaccharides (LPS), peptidoglycans, and surface proteins. Endocytic PRRs on phagocytes reflect these molecular patterns, with C-type lectin receptors binding to mannans and β-glucans, and scavenger receptors binding to LPS.
Upon endocytic PRR binding, actin-myosin cytoskeletal rearrangement adjacent to the plasma membrane occurs in a way that endocytoses the plasma membrane containing the PRR-PAMP complex, and the microbe. Phosphatidylinositol and Vps34-Vps15-Beclin1 signalling pathways have been implicated to traffic the endocytosed phagosome to intracellular lysosomes, where fusion of the phagosome and the lysosome produces a phagolysosome. The reactive oxygen species, superoxides and hypochlorite bleach within the phagolysosomes then kill microbes inside the phagocyte.
Phagocytic efficacy can be enhanced by opsonization. Plasma derived complement C3b and antibodies that exude into the inflamed tissue during the vascular phase bind to and coat the microbial antigens. As well as endocytic PRRs, phagocytes also express opsonin receptors Fc receptor and complement receptor 1 (CR1), which bind to antibodies and C3b, respectively. The co-stimulation of endocytic PRR and opsonin receptor increases the efficacy of the phagocytic process, enhancing the lysosomal elimination of the infective agent.
Cell-derived mediators
* non-exhaustive list
| Name | Type | Source | Description |
|---|---|---|---|
| Lysosome granules | Enzymes | Granulocytes | These cells contain a large variety of enzymes that perform a number of functions. Granules can be classified as either specific or azurophilic depending upon the contents, and are able to break down a number of substances, some of which may be plasma-derived proteins that allow these enzymes to act as inflammatory mediators. |
| Histamine | Monoamine | Mast cells and basophils | Stored in preformed granules, histamine is released in response to a number of stimuli. It causes arteriole dilation, increased venous permeability, and a wide variety of organ-specific effects. |
| IFN-γ | Cytokine | T-cells, NK cells | Antiviral, immunoregulatory, and anti-tumour properties. This interferon was originally called macrophage-activating factor, and is especially important in the maintenance of chronic inflammation. |
| IL-8 | Chemokine | Primarily macrophages | Activation and chemoattraction of neutrophils, with a weak effect on monocytes and eosinophils. |
| Leukotriene B4 | Eicosanoid | Leukocytes, cancer cells | Able to mediate leukocyte adhesion and activation, allowing them to bind to the endothelium and migrate across it. In neutrophils, it is also a potent chemoattractant, and is able to induce the formation of reactive oxygen species and the release of lysosomal enzymes by these cells. |
| LTC4, LTD4 | Eicosanoid | eosinophils, mast cells, macrophages | These three Cysteine-containing leukotrienes contract lung airways, increase micro-vascular permeability, stimulate mucus secretion, and promote eosinophil-based inflammation in the lung, skin, nose, eye, and other tissues. |
| 5-oxo-eicosatetraenoic acid | Eicosanoid | leukocytes, cancer cells | Potent stimulator of neutrophil chemotaxis, lysosome enzyme release, and reactive oxygen species formation; monocyte chemotaxis; and with even greater potency eosinophil chemotaxis, lysosome enzyme release, and reactive oxygen species formation. |
| 5-HETE | Eicosanoid | Leukocytes | Metabolic precursor to 5-Oxo-eicosatetraenoic acid, it is a less potent stimulator of neutrophil chemotaxis, lysosome enzyme release, and reactive oxygen species formation; monocyte chemotaxis; and eosinophil chemotaxis, lysosome enzyme release, and reactive oxygen species formation. |
| Prostaglandins | Eicosanoid | Mast cells | A group of lipids that can cause vasodilation, fever, and pain. |
| Nitric oxide | Soluble gas | Macrophages, endothelial cells, some neurons | Potent vasodilator, relaxes smooth muscle, reduces platelet aggregation, aids in leukocyte recruitment, direct antimicrobial activity in high concentrations. |
| TNF-α and IL-1 | Cytokines | Primarily macrophages | Both affect a wide variety of cells to induce many similar inflammatory reactions: fever, production of cytokines, endothelial gene regulation, chemotaxis, leukocyte adherence, activation of fibroblasts. Responsible for the systemic effects of inflammation, such as loss of appetite and increased heart rate. TNF-α inhibits osteoblast differentiation. |
| Tryptase | Enzymes | Mast Cells | This serine protease is believed to be exclusively stored in mast cells and secreted, along with histamine, during mast cell activation.[16][17][18] |
Morphologic patterns
Specific patterns of acute and chronic inflammation are seen during particular situations that arise in the body, such as when inflammation occurs on an epithelial surface, or pyogenic bacteria are involved.
- Granulomatous inflammation: Characterised by the formation of granulomas, they are the result of a limited but diverse number of diseases, which include among others tuberculosis, leprosy, sarcoidosis, and syphilis.
- Fibrinous inflammation: Inflammation resulting in a large increase in vascular permeability allows fibrin to pass through the blood vessels. If an appropriate procoagulative stimulus is present, such as cancer cells,[8] a fibrinous exudate is deposited. This is commonly seen in serous cavities, where the conversion of fibrinous exudate into a scar can occur between serous membranes, limiting their function. The deposit sometimes forms a pseudomembrane sheet. During inflammation of the intestine (Pseudomembranous colitis), pseudomembranous tubes can be formed.
- Purulent inflammation: Inflammation resulting in large amount of pus, which consists of neutrophils, dead cells, and fluid. Infection by pyogenic bacteria such as staphylococci is characteristic of this kind of inflammation. Large, localised collections of pus enclosed by surrounding tissues are called abscesses.
- Serous inflammation: Characterised by the copious effusion of non-viscous serous fluid, commonly produced by mesothelial cells of serous membranes, but may be derived from blood plasma. Skin blisters exemplify this pattern of inflammation.
- Ulcerative inflammation: Inflammation occurring near an epithelium can result in the necrotic loss of tissue from the surface, exposing lower layers. The subsequent excavation in the epithelium is known as an ulcer.
Inflammatory disorders
.png)
Inflammatory abnormalities are a large group of disorders that underlie a vast variety of human diseases. The immune system is often involved with inflammatory disorders, demonstrated in both allergic reactions and some myopathies, with many immune system disorders resulting in abnormal inflammation. Non-immune diseases with causal origins in inflammatory processes include cancer, atherosclerosis, and ischemic heart disease.[8]
Examples of disorders associated with inflammation include:
- Acne vulgaris
- Asthma
- Autoimmune diseases
- Autoinflammatory diseases
- Celiac disease
- Chronic prostatitis
- Colitis
- Diverticulitis
- Glomerulonephritis
- Hidradenitis suppurativa
- Hypersensitivities
- Inflammatory bowel diseases
- Interstitial cystitis
- Lichen planus
- Mast Cell Activation Syndrome
- Mastocytosis
- Otitis
- Pelvic inflammatory disease
- Reperfusion injury
- Rheumatic fever
- Rheumatoid arthritis
- Rhinitis
- Sarcoidosis
- Transplant rejection
- Vasculitis
Atherosclerosis
Atherosclerosis, formerly considered a bland lipid storage disease, actually involves an ongoing inflammatory response. Recent advances in basic science have established a fundamental role for inflammation in mediating all stages of this disease from initiation through progression and, ultimately, the thrombotic complications of atherosclerosis. These new findings provide important links between risk factors and the mechanisms of atherogenesis. Clinical studies have shown that this emerging biology of inflammation in atherosclerosis applies directly to human patients. Elevation in markers of inflammation predicts outcomes of patients with acute coronary syndromes, independently of myocardial damage. In addition, low-grade chronic inflammation, as indicated by levels of the inflammatory marker C-reactive protein, prospectively defines risk of atherosclerotic complications, thus adding to prognostic information provided by traditional risk factors. Moreover, certain treatments that reduce coronary risk also limit inflammation. In the case of lipid lowering with statins, this anti-inflammatory effect does not appear to correlate with reduction in low-density lipoprotein levels. These new insights into inflammation in atherosclerosis not only increase our understanding of this disease but also have practical clinical applications in risk stratification and targeting of therapy for this scourge of growing worldwide importance.[19]
Allergy
An allergic reaction, formally known as type 1 hypersensitivity, is the result of an inappropriate immune response triggering inflammation, vasodilation, and nerve irritation. A common example is hay fever, which is caused by a hypersensitive response by mast cells to allergens. Pre-sensitised mast cells respond by degranulating, releasing vasoactive chemicals such as histamine. These chemicals propagate an excessive inflammatory response characterised by blood vessel dilation, production of pro-inflammatory molecules, cytokine release, and recruitment of leukocytes.[8] Severe inflammatory response may mature into a systemic response known as anaphylaxis.
Myopathies
Inflammatory myopathies are caused by the immune system inappropriately attacking components of muscle, leading to signs of muscle inflammation. They may occur in conjunction with other immune disorders, such as systemic sclerosis, and include dermatomyositis, polymyositis, and inclusion body myositis.[8]
Leukocyte defects
Due to the central role of leukocytes in the development and propagation of inflammation, defects in leukocyte functionality often result in a decreased capacity for inflammatory defense with subsequent vulnerability to infection.[8] Dysfunctional leukocytes may be unable to correctly bind to blood vessels due to surface receptor mutations, digest bacteria (Chédiak–Higashi syndrome), or produce microbicides (chronic granulomatous disease). In addition, diseases affecting the bone marrow may result in abnormal or few leukocytes.
Pharmacological
Certain drugs or exogenous chemical compounds are known to affect inflammation. Vitamin A deficiency causes an increase in inflammatory responses,[20] and anti-inflammatory drugs work specifically by inhibiting the enzymes that produce inflammatory eicosanoids. Certain illicit drugs such as cocaine and ecstasy may exert some of their detrimental effects by activating transcription factors intimately involved with inflammation (e.g. NF-κB).[21][22]
Cancer
Inflammation orchestrates the microenvironment around tumours, contributing to proliferation, survival and migration.[23] Cancer cells use selectins, chemokines and their receptors for invasion, migration and metastasis.[24] On the other hand, many cells of the immune system contribute to cancer immunology, suppressing cancer.[25] Molecular intersection between receptors of steroid hormones, which have important effects on cellular development, and transcription factors that play key roles in inflammation, such as NF-κB, may mediate some of the most critical effects of inflammatory stimuli on cancer cells.[26] This capacity of a mediator of inflammation to influence the effects of steroid hormones in cells, is very likely to affect carcinogenesis on the one hand; on the other hand, due to the modular nature of many steroid hormone receptors, this interaction may offer ways to interfere with cancer progression, through targeting of a specific protein domain in a specific cell type. Such an approach may limit side effects that are unrelated to the tumor of interest, and may help preserve vital homeostatic functions and developmental processes in the organism.
According to a review of 2009, recent data suggests that cancer-related inflammation (CRI) may lead to accumulation of random genetic alterations in cancer cells.[27]
Importance of inflammation in cancer
In 1863, Rudolf Virchow hypothesized that the origin of cancer was at sites of chronic inflammation.[28][29] At present, chronic inflammation is estimated to contribute to approximately 15% to 25% of human cancers.[30][29][31][32]
Mediators and DNA damage in cancer
An inflammatory mediator is a messenger that acts on blood vessels and/or cells to promote an inflammatory response.[33] Inflammatory mediators that contribute to neoplasia include prostaglandins, inflammatory cytokines such as IL-1β, TNF-α, IL-6 and IL-15 and chemokines such as IL-8 and GRO-alpha.[34][29] These inflammatory mediators, and others, orchestrate an environment that fosters proliferation and survival.[28][34]
Inflammation also causes DNA damages due to the induction of reactive oxygen species (ROS) by various intracellular inflammatory mediators.[28][34][29] In addition, leukocytes and other phagocytic cells attracted to the site of inflammation induce DNA damages in proliferating cells through their generation of ROS and reactive nitrogen species (RNS). ROS and RNS are normally produced by these cells to fight infection.[28] ROS, alone, cause more than 20 types of DNA damage.[35] Oxidative DNA damages cause both mutations[36] and epigenetic alterations.[37][29][31] RNS also cause mutagenic DNA damages.[38]
A normal cell may undergo carcinogenesis to become a cancer cell if it is frequently subjected to DNA damage during long periods of chronic inflammation. DNA damages may cause genetic mutations due to inaccurate repair. In addition, mistakes in the DNA repair process may cause epigenetic alterations.[29][34][31] Mutations and epigenetic alterations that are replicated and provide a selective advantage during somatic cell proliferation may be carcinogenic.
Genome-wide analyses of human cancer tissues reveal that a single typical cancer cell may possess roughly 100 mutations in coding regions, 10-20 of which are “driver mutations” that contribute to cancer development.[29] However, chronic inflammation also causes epigenetic changes such as DNA methylations, that are often more common than mutations. Typically, several hundreds to thousands of genes are methylated in a cancer cell (see DNA methylation in cancer). Sites of oxidative damage in chromatin can recruit complexes that contain DNA methyltransferases (DNMTs), a histone deacetylase (SIRT1), and a histone methyltransferase (EZH2), and thus induce DNA methylation.[29][39][40] DNA methylation of a CpG island in a promoter region may cause silencing of its downstream gene (see CpG site and regulation of transcription in cancer). DNA repair genes, in particular, are frequently inactivated by methylation in various cancers (see hypermethylation of DNA repair genes in cancer). A 2018 report[41] evaluated the relative importance of mutations and epigenetic alterations in progression to two different types of cancer. This report showed that epigenetic alterations were much more important than mutations in generating gastric cancers (associated with inflammation).[42] However, mutations and epigenetic alterations were of roughly equal importance in generating esophageal squamous cell cancers (associated with tobacco chemicals and acetaldehyde, a product of alcohol metabolism).
HIV and AIDS
It has long been recognized that infection with HIV is characterized not only by development of profound immunodeficiency but also by sustained inflammation and immune activation.[43][44][45] A substantial body of evidence implicates chronic inflammation as a critical driver of immune dysfunction, premature appearance of aging-related diseases, and immune deficiency.[43][46] Many now regard HIV infection not only as an evolving virus-induced immunodeficiency but also as chronic inflammatory disease.[47] Even after the introduction of effective antiretroviral therapy (ART) and effective suppression of viremia in HIV-infected individuals, chronic inflammation persists. Animal studies also support the relationship between immune activation and progressive cellular immune deficiency: SIVsm infection of its natural nonhuman primate hosts, the sooty mangabey, causes high-level viral replication but limited evidence of disease.[48][49] This lack of pathogenicity is accompanied by a lack of inflammation, immune activation and cellular proliferation. In sharp contrast, experimental SIVsm infection of rhesus macaque produces immune activation and AIDS-like disease with many parallels to human HIV infection.[50]
Delineating how CD4 T cells are depleted and how chronic inflammation and immune activation are induced lies at the heart of understanding HIV pathogenesis––one of the top priorities for HIV research by the Office of AIDS Research, National Institutes of Health. Recent studies demonstrated that caspase-1-mediated pyroptosis, a highly inflammatory form of programmed cell death, drives CD4 T-cell depletion and inflammation by HIV.[51][52][53] These are the two signature events that propel HIV disease progression to AIDS. Pyroptosis appears to create a pathogenic vicious cycle in which dying CD4 T cells and other immune cells (including macrophages and neutrophils) release inflammatory signals that recruit more cells into the infected lymphoid tissues to die. The feed-forward nature of this inflammatory response produces chronic inflammation and tissue injury.[54] Identifying pyroptosis as the predominant mechanism that causes CD4 T-cell depletion and chronic inflammation, provides novel therapeutic opportunities, namely caspase-1 which controls the pyroptotic pathway. In this regard, pyroptosis of CD4 T cells and secretion of pro-inflmammatory cytokines such as IL-1β and IL-18 can be blocked in HIV-infected human lymphoid tissues by addition of the caspase-1 inhibitor VX-765,[51] which has already proven to be safe and well tolerated in phase II human clinical trials.[55] These findings could propel development of an entirely new class of “anti-AIDS” therapies that act by targeting the host rather than the virus. Such agents would almost certainly be used in combination with ART. By promoting “tolerance” of the virus instead of suppressing its replication, VX-765 or related drugs may mimic the evolutionary solutions occurring in multiple monkey hosts (e.g. the sooty mangabey) infected with species-specific lentiviruses that have led to a lack of disease, no decline in CD4 T-cell counts, and no chronic inflammation.
Resolution of inflammation
The inflammatory response must be actively terminated when no longer needed to prevent unnecessary "bystander" damage to tissues.[8] Failure to do so results in chronic inflammation, and cellular destruction. Resolution of inflammation occurs by different mechanisms in different tissues. Mechanisms that serve to terminate inflammation include:[8][56]
- Short half-life of inflammatory mediators in vivo.
- Production and release of transforming growth factor (TGF) beta from macrophages[57][58][59]
- Production and release of interleukin 10 (IL-10)[60]
- Production of anti-inflammatory specialized proresolving mediators, i.e. lipoxins, resolvins, maresins, and neuroprotectins[61][62]
- Downregulation of pro-inflammatory molecules, such as leukotrienes.
- Upregulation of anti-inflammatory molecules such as the interleukin 1 receptor antagonist or the soluble tumor necrosis factor receptor (TNFR)
- Apoptosis of pro-inflammatory cells[63]
- Desensitization of receptors.
- Increased survival of cells in regions of inflammation due to their interaction with the extracellular matrix (ECM)[64][65]
- Downregulation of receptor activity by high concentrations of ligands
- Cleavage of chemokines by matrix metalloproteinases (MMPs) might lead to production of anti-inflammatory factors.[66]
| “ | Acute inflammation normally resolves by mechanisms that have remained somewhat elusive. Emerging evidence now suggests that an active, coordinated program of resolution initiates in the first few hours after an inflammatory response begins. After entering tissues, granulocytes promote the switch of arachidonic acid–derived prostaglandins and leukotrienes to lipoxins, which initiate the termination sequence. Neutrophil recruitment thus ceases and programmed death by apoptosis is engaged. These events coincide with the biosynthesis, from omega-3 polyunsaturated fatty acids, of resolvins and protectins, which critically shorten the period of neutrophil infiltration by initiating apoptosis. As a consequence, apoptotic neutrophils undergo phagocytosis by macrophages, leading to neutrophil clearance and release of anti-inflammatory and reparative cytokines such as transforming growth factor-β1. The anti-inflammatory program ends with the departure of macrophages through the lymphatics.[67] | ” |
| — Charles Serhan | ||
Connection to depression
There is evidence for a link between inflammation and depression.[68] Inflammatory processes can be triggered by negative cognitions or their consequences, such as stress, violence, or deprivation. Thus, negative cognitions can cause inflammation that can, in turn, lead to depression.[69][70] In addition there is increasing evidence that inflammation can cause depression because of the increase of cytokines, setting the brain into a "sickness mode".[71] Classical symptoms of being physically sick like lethargy show a large overlap in behaviors that characterize depression. Levels of cytokines tend to increase sharply during the depressive episodes of people with bipolar disorder and drop off during remission.[72] Furthermore, it has been shown in clinical trials that anti-inflammatory medicines taken in addition to antidepressants not only significantly improves symptoms but also increases the proportion of subjects positively responding to treatment.[73] Inflammations that lead to serious depression could be caused by common infections such as those caused by a virus, bacteria or even parasites.[74]
Systemic effects
An infectious organism can escape the confines of the immediate tissue via the circulatory system or lymphatic system, where it may spread to other parts of the body. If an organism is not contained by the actions of acute inflammation it may gain access to the lymphatic system via nearby lymph vessels. An infection of the lymph vessels is known as lymphangitis, and infection of a lymph node is known as lymphadenitis. When lymph nodes cannot destroy all pathogens, the infection spreads further. A pathogen can gain access to the bloodstream through lymphatic drainage into the circulatory system.
When inflammation overwhelms the host, systemic inflammatory response syndrome is diagnosed. When it is due to infection, the term sepsis is applied, with the terms bacteremia being applied specifically for bacterial sepsis and viremia specifically to viral sepsis. Vasodilation and organ dysfunction are serious problems associated with widespread infection that may lead to septic shock and death.
Acute-phase proteins
Inflammation also induces high systemic levels of acute-phase proteins. In acute inflammation, these proteins prove beneficial; however, in chronic inflammation they can contribute to amyloidosis.[8] These proteins include C-reactive protein, serum amyloid A, and serum amyloid P, which cause a range of systemic effects including:[8]
- Fever
- Increased blood pressure
- Decreased sweating
- Malaise
- Loss of appetite
- Somnolence
Leukocyte numbers
Inflammation often affects the numbers of leukocytes present in the body:
- Leukocytosis is often seen during inflammation induced by infection, where it results in a large increase in the amount of leukocytes in the blood, especially immature cells. Leukocyte numbers usually increase to between 15 000 and 20 000 cells per microliter, but extreme cases can see it approach 100 000 cells per microliter.[8] Bacterial infection usually results in an increase of neutrophils, creating neutrophilia, whereas diseases such as asthma, hay fever, and parasite infestation result in an increase in eosinophils, creating eosinophilia.[8]
- Leukopenia can be induced by certain infections and diseases, including viral infection, Rickettsia infection, some protozoa, tuberculosis, and some cancers.[8]
Systemic inflammation and obesity
With the discovery of interleukins (IL), the concept of systemic inflammation developed. Although the processes involved are identical to tissue inflammation, systemic inflammation is not confined to a particular tissue but involves the endothelium and other organ systems.
Chronic inflammation is widely observed in obesity.[75][76] Obese people commonly have many elevated markers of inflammation, including:[77][78]
- IL-8 (Interleukin-8)
- IL-18 (Interleukin-18)[79][80]
- TNF-α (Tumor necrosis factor-alpha)[79][80]
- CRP (C-reactive protein)[79][80]
- Insulin[79][80]
- Blood glucose[79][80]
- Leptin[79][80]
Low-grade chronic inflammation is characterized by a two- to threefold increase in the systemic concentrations of cytokines such as TNF-α, IL-6, and CRP.[81] Waist circumference correlates significantly with systemic inflammatory response.[82]
Loss of white adipose tissue reduces levels of inflammation markers.[75] The association of systemic inflammation with insulin resistance and type 2 diabetes, and with atherosclerosis is under preliminary research, although rigorous clinical trials have not been conducted to confirm such relationships.[83]
C-reactive protein (CRP) is generated at a higher level in obese people, and may increase the risk for cardiovascular diseases.[84]
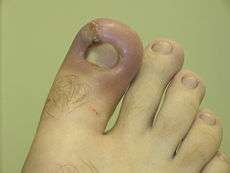
Outcomes
.jpg)
The outcome in a particular circumstance will be determined by the tissue in which the injury has occurred and the injurious agent that is causing it. Here are the possible outcomes to inflammation:[8]
- Resolution
The complete restoration of the inflamed tissue back to a normal status. Inflammatory measures such as vasodilation, chemical production, and leukocyte infiltration cease, and damaged parenchymal cells regenerate. In situations where limited or short-lived inflammation has occurred this is usually the outcome. - Fibrosis
Large amounts of tissue destruction, or damage in tissues unable to regenerate, cannot be regenerated completely by the body. Fibrous scarring occurs in these areas of damage, forming a scar composed primarily of collagen. The scar will not contain any specialized structures, such as parenchymal cells, hence functional impairment may occur. - Abscess formation
A cavity is formed containing pus, an opaque liquid containing dead white blood cells and bacteria with general debris from destroyed cells. - Chronic inflammation
In acute inflammation, if the injurious agent persists then chronic inflammation will ensue. This process, marked by inflammation lasting many days, months or even years, may lead to the formation of a chronic wound. Chronic inflammation is characterised by the dominating presence of macrophages in the injured tissue. These cells are powerful defensive agents of the body, but the toxins they release (including reactive oxygen species) are injurious to the organism's own tissues as well as invading agents. As a consequence, chronic inflammation is almost always accompanied by tissue destruction.
Examples
Inflammation is usually indicated by adding the suffix "itis", as shown below. However, some conditions such as asthma and pneumonia do not follow this convention. More examples are available at list of types of inflammation.
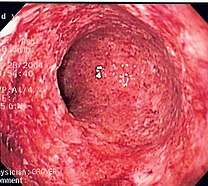
 Acute appendicitis
Acute appendicitis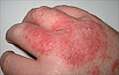 Acute dermatitis
Acute dermatitis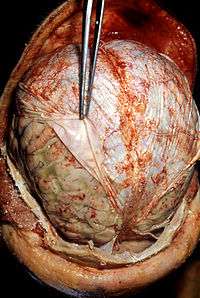 Acute infective meningitis
Acute infective meningitis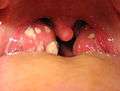 Acute tonsillitis
Acute tonsillitis
Diet and inflammation
The Dietary Inflammatory Index (DII) is a score (number) that describes the potential of diet to modulate systemic inflammation within the body. As stated chronic inflammation is linked to most chronic diseases including arthritis, many types of cancer, cardiovascular diseases, inflammatory bowel diseases, and diabetes.
Exercise and inflammation
Exercise-induced acute inflammation
Acute inflammation of the muscle cells, as understood in exercise physiology,[85] can result after induced eccentric and concentric muscle training. Participation in eccentric training and conditioning, including resistance training and activities that emphasize eccentric lengthening of the muscle including downhill running on a moderate to high incline can result in considerable soreness within 24 to 48 hours, even though blood lactate levels, previously thought to cause muscle soreness, were much higher with level running. This delayed onset muscle soreness (DOMS) results from structural damage to the contractile filaments and z-disks, which has been noted especially in marathon runners whose muscle fibers revealed remarkable damage to the muscle fibers after both training and marathon competition [86]. The onset and timing of this gradient damage to the muscle parallels the degree of muscle soreness experienced by the runners.
Z-disks are the point of contact for the contractile proteins. They provide structural support for transmission of force when muscle fibers are activated to shorten. However, in marathon runners and those who subscribe to the overload principle to enhance their muscles, show moderate Z-disk streaming and major disruption of thick and thin filaments in parallel groups of sarcomeres as a result of the force of eccentric actions or stretching of tightened muscle fibers.
This disruption of muscle fibers triggers white blood cells to increase following induced muscle soreness, leading to the inflammatory response observation from induced muscle soreness. Elevations in plasma enzymes, myoglobinemia, and abnormal muscle histology and ultrastructure are concluded to be associated with inflammatory response. High tension in the contractile-elastic system of muscle results in structural damage to the muscle fiber and plasmalemma and its epimysium, perimysium, and/or endomysium. The mysium damage disrupts calcium homeostasis in injured fibers and fiber bundles, resulting in necrosis that peaks about 48 hours after exercise. The products of macrophage activity and intracellular contents (such as histamines, kinins, and K+) accumulate outside cells. These substances then stimulate free nerve endings in the muscle; a process that appears accentuated by eccentric exercise, in which large forces are distributed over a relatively small cross-sectional area of the muscle[86] .
Post-inflammatory muscle growth and repair
There is a known relationship between inflammation and muscle growth.[87] For instance, high doses of anti-inflammatory medicines (e.g., NSAIDs) are able to blunt muscle growth.[88][89] Cold therapy has been shown to negatively affect muscle growth as well. Reducing inflammation results in decreased macrophage activity and lower levels of IGF-1[90] Acute effects of cold therapy on training adaptations show reduced satellite cell proliferation.[91] Long term effects include less muscular hypertrophy and an altered cell structure of muscle fibers.[92]
It has been further theorized that the acute localized inflammatory responses to muscular contraction during exercise, as described above, are a necessary precursor to muscle growth.[93] As a response to muscular contractions, the acute inflammatory response initiates the breakdown and removal of damaged muscle tissue.[94] Muscles can synthesize cytokines in response to contractions,[95][96][97] such that the cytokines interleukin-1 beta (IL-1β), TNF-α, and IL-6 are expressed in skeletal muscle up to 5 days after exercise.[94]
In particular, the increase in levels of IL-6 (interleukin 6), a myokine, can reach up to one hundred times that of resting levels.[97] Depending on volume, intensity, and other training factors, the IL-6 increase associated with training initiates about 4 hours after resistance training and remains elevated for up to 24 hours.[98][99][100]
These acute increases in cytokines, as a response to muscle contractions, help initiate the process of muscle repair and growth by activating satellite cells within the inflamed muscle. Satellite cells are crucial for skeletal muscle adaptation to exercise.[101] They contribute to hypertrophy by providing new myonuclei and repair damaged segments of mature myofibers for successful regeneration following injury- or exercise-induced muscle damage;[102][103][104] high-level powerlifters can have up to 100% more satellite cells than untrained controls.[105][106]
A rapid and transient localization of the IL-6 receptor and increased IL-6 expression occurs in satellite cells following contractions.[98] IL-6 has been shown to mediate hypertrophic muscle growth both in vitro and in vivo.[101] Unaccustomed exercise can increase IL-6 by up to sixfold at 5 hours post-exercise and threefold 8 days after exercise.[107] Also telling is the fact that NSAIDs can decrease satellite cell response to exercise,[88] thereby reducing exercise-induced protein synthesis.[89]
The increase in cytokines (myokines) after resistance exercise coincides with the decrease in levels of myostatin, a protein that inhibits muscle differentiation and growth.[100] The cytokine response to resistance exercise and moderate-intensity running occur differently, with the latter causing a more prolonged response, especially at the 12–24 hour mark.[100]
Developing research has demonstrated that many of the benefits of exercise are mediated through the role of skeletal muscle as an endocrine organ. That is, contracting muscles release multiple substances known as myokines, including but not limited to those cited in the above description, which promote the growth of new tissue, tissue repair, and various anti-inflammatory functions, which in turn reduce the risk of developing various inflammatory diseases. The new view that muscle is an endocrine organ is transforming our understanding of exercise physiology and with it, of the role of inflammation in adaptation to stress.[108]
Chronic inflammation and muscle loss
Both chronic and extreme inflammation are associated with disruptions of anabolic signals initiating muscle growth. Chronic inflammation has been implicated as part of the cause of the muscle loss that occurs with aging.[87][109] Increased protein levels of myostatin have been described in patients with diseases characterized by chronic low-grade inflammation.[110] Increased levels of TNF-α can suppress the AKT/mTOR pathway, a crucial pathway for regulating skeletal muscle hypertrophy,[111] thereby increasing muscle catabolism.[112][113][114] Cytokines may antagonize the anabolic effects of insulin-like growth factor 1 (IGF-1).[115][116] In the case of sepsis, an extreme whole body inflammatory state, the synthesis of both myofibrillar and sarcoplasmic proteins are inhibited, with the inhibition taking place preferentially in fast-twitch muscle fibers.[115][117] Sepsis is also able to prevent leucine from stimulating muscle protein synthesis.[95] In animal models, when inflammation is created, mTOR loses its ability to be stimulated by muscle growth.[118]
Exercise as a treatment for inflammation
Regular physical activity is reported to decrease markers of inflammation,[119][120][121] although the correlation is imperfect and seems to reveal differing results contingent upon training intensity. For instance, while baseline measurements of circulating inflammatory markers do not seem to differ greatly between healthy trained and untrained adults,[122][123] long-term training may help reduce chronic low-grade inflammation.[124] On the other hand, levels of the anti-inflammatory myokine IL-6 (interleukin 6) remained elevated longer into the recovery period following an acute bout of exercise in patients with inflammatory diseases, relative to the recovery of healthy controls.[124] It may well be that low-intensity training can reduce resting pro-inflammatory markers (CRP, IL-6), while moderate-intensity training has milder and less-established anti-inflammatory benefits.[122][125][126][127] There is a strong relationship between exhaustive exercise and chronic low-grade inflammation.[128] Marathon running may enhance IL-6 levels as much as 100 times over normal and increases total leuckocyte count and neturophil mobilization.[128]
Regarding the above, IL-6 had previously been classified as a proinflammatory cytokine. Therefore, it was first thought that the exercise-induced IL-6 response was related to muscle damage.[129] However, it has become evident that eccentric exercise is not associated with a larger increase in plasma IL-6 than exercise involving concentric “nondamaging” muscle contractions. This finding clearly demonstrates that muscle damage is not required to provoke an increase in plasma IL-6 during exercise. As a matter of fact, eccentric exercise may result in a delayed peak and a much slower decrease of plasma IL-6 during recovery.[130]
Recent work has shown that both upstream and downstream signalling pathways for IL-6 differ markedly between myocytes and macrophages. It appears that unlike IL-6 signalling in macrophages, which is dependent upon activation of the NFκB signalling pathway, intramuscular IL-6 expression is regulated by a network of signalling cascades, including the Ca2+/NFAT and glycogen/p38 MAPK pathways. Thus, when IL-6 is signalling in monocytes or macrophages, it creates a pro-inflammatory response, whereas IL-6 activation and signalling in muscle is totally independent of a preceding TNF-response or NFκB activation, and is anti-inflammatory.[131]
Several studies show that markers of inflammation are reduced following longer-term behavioural changes involving both reduced energy intake and a regular program of increased physical activity, and that, in particular, IL-6 was miscast as an inflammatory marker. For example, the anti-inflammatory effects of IL-6 have been demonstrated by IL-6 stimulating the production of the classical anti-inflammatory cytokines IL-1ra and IL-10.[131] As such, individuals pursuing exercise as a means to treat the causal factors underlying chronic inflammation are pursuing a course of action strongly supported by current research, as an inactive lifestyle is strongly associated with the development and progression of multiple inflammatory diseases. Note that cautions regarding over-exertion may apply in certain cases, as discussed above, though this concern rarely applies to the general population.
Signal-to-noise theory
Given that localized acute inflammation is a necessary component for muscle growth, and that chronic low-grade inflammation is associated with a disruption of anabolic signals initiating muscle growth, it has been theorized that a signal-to-noise model may best describe the relationship between inflammation and muscle growth.[132] By keeping the "noise" of chronic inflammation to a minimum, the localized acute inflammatory response signals a stronger anabolic response than could be achieved with higher levels of chronic inflammation.
See also
References
- Ferrero-Miliani L, Nielsen OH, Andersen PS, Girardin SE; Nielsen; Andersen; Girardin (February 2007). "Chronic inflammation: importance of NOD2 and NALP3 in interleukin-1beta generation". Clin. Exp. Immunol. 147 (2): 061127015327006––. doi:10.1111/j.1365-2249.2006.03261.x. PMC 1810472. PMID 17223962.CS1 maint: multiple names: authors list (link)
- Abbas A.B.; Lichtman A.H. (2009). "Ch.2 Innate Immunity". In Saunders (Elsevier) (ed.). Basic Immunology. Functions and disorders of the immune system (3rd ed.). ISBN 978-1-4160-4688-2.
- Hall, John (2011). Guyton and Hall textbook of medical physiology (12th ed.). Philadelphia, Pa.: Saunders/Elsevier. p. 428. ISBN 978-1-4160-4574-8.
- Granger, D. Neil; Senchenkova, Elena (2010). "Leukocyte–Endothelial Cell Adhesion". Inflammation and the Microcirculation. Integrated Systems Physiology—From Cell to Function. Morgan & Claypool Life Sciences.
- Piira, Olli-Pekka; Miettinen, Johanna A.; Hautala, Arto J.; Huikuri, Heikki V.; Tulppo, Mikko P. (2013). "Physiological responses to emotional excitement in healthy subjects and patients with coronary artery disease". Autonomic Neuroscience. 177 (2): 280–5. doi:10.1016/j.autneu.2013.06.001. PMID 23916871.
- Stedman's Medical Dictionary (Twenty-fifth ed.). Williams & Wilkins. 1990.
- Rather, L. J. (1971). "Disturbance of function (functio laesa): the legendary fifth cardinal sign of inflammation, added by Galen to the four cardinal signs of Celsus". Bull N Y Acad Med. 47 (3): 303–322. PMC 1749862. PMID 5276838.
- Cotran; Kumar, Collins (1998). Robbins Pathologic Basis of Disease. Philadelphia: W.B Saunders Company. ISBN 978-0-7216-7335-6.
- Kumar, Rukmini; Clermont, Gilles; Vodovotz, Yoram; Chow, Carson C. (21 September 2004). "The dynamics of acute inflammation". Journal of Theoretical Biology. 230 (2): 145–155. arXiv:q-bio/0404034. Bibcode:2004PhDT.......405K. doi:10.1016/j.jtbi.2004.04.044. PMID 15321710.
- Parakrama Chandrasoma; Clive R. Taylor (2005). "Part A. "General Pathology", Section II. "The Host Response to Injury", Chapter 3. "The Acute Inflammatory Response", sub-section "Cardinal Clinical Signs"". Concise Pathology (3rd ed.). McGraw-Hill. ISBN 978-0-8385-1499-3. OCLC 150148447. Retrieved 5 November 2008.
- Werner, Ruth (2009). A massage Therapist Guide to Pathology (4th ed.). Wolters Kluwer. ISBN 978-0781769198.
- Vogel, Wolfgang H.; Berke, Andreas (2009). Brief History of Vision and Ocular Medicine. Kugler Publications. p. 97. ISBN 978-90-6299-220-1.
- Porth, Carol (2007). Essentials of pahtophysiology: concepts of altered health states. Hagerstown, MD: Lippincott Williams & Wilkins. p. 270. ISBN 978-0-7817-7087-3.
- Dormandy, Thomas (2006). The worst of evils: man's fight against pain. New Haven, Conn: Yale University Press. p. 22. ISBN 978-0-300-11322-8.
- Herrington, Simon (2014). Muir's Textbook of Pathology (15th ed.). CRC Press. p. 59. ISBN 978-1444184990.
- Cevikbas, Ferda; Kempkes, Cordula; Buhl, Timo; Mess, Christian; Buddenkotte, Joerg; Steinhoff, Martin (2014). Carstens, E.; Akiyama, Tasuku (eds.). Itch: Mechanisms and Treatment. Frontiers in Neuroscience. Boca Raton (FL): CRC Press/Taylor & Francis. ISBN 9781466505438. PMID 24830021.
- Caughey, George H. (1 June 2007). "Mast cell tryptases and chymases in inflammation and host defense". Immunological Reviews. 217 (1): 141–154. doi:10.1111/j.1600-065x.2007.00509.x. PMC 2275918. PMID 17498057.
- Caughey, George H. (5 May 2016). "Mast cell proteases as pharmacological targets". European Journal of Pharmacology. Pharmacological modulation of Mast cells and Basophils. 778: 44–55. doi:10.1016/j.ejphar.2015.04.045. PMC 4636979. PMID 25958181.
- Libby, P (19–26 December 2002). "Inflammation in atherosclerosis". Nature. 420 (6917): 868–74. Bibcode:2002Natur.420..868L. doi:10.1038/nature01323. PMID 12490960.
- Wiedermann U, et al. (1996). "Vitamin A deficiency increases inflammatory responses". Scand J Immunol. 44 (6): 578–584. doi:10.1046/j.1365-3083.1996.d01-351.x. PMID 8972739.
- Hargrave, B. Y.; Tiangco, D. A.; Lattanzio, F. A.; Beebe, S. J. (2003). "Cocaine, not morphine, causes the generation of reactive oxygen species and activation of NF-κB in transiently cotransfected heart cells". Cardiovasc Toxicol. 3 (2): 141–151. doi:10.1385/CT:3:2:141. PMID 14501032.
- Montiel-Duarte, C.; Ansorena, E.; López-Zabalza, M. J.; Cenarruzabeitia, E.; Iraburu, M. J. (2004). "Role of reactive oxygen species, glutathione and NF-κB in apoptosis induced by 3,4-methylenedioxymethamphetamine ("Ecstasy") on hepatic stellate cells". Biochem Pharmacol. 67 (6): 1025–33. doi:10.1016/j.bcp.2003.10.020. PMID 15006539.
- Hendrik Ungefroren; Susanne Sebens; Daniel Seidl; Hendrik Lehnert; Ralf Haas (2011). "Interaction of tumor cells with the microenvironment". Cell Communication and Signaling. 9 (18). doi:10.1186/1478-811X-9-18. PMC 3180438. PMID 21914164.
- Coussens, L. M.; Werb, Z. (2002). "Inflammation and cancer". Nature. 420 (6917): 860–7. Bibcode:2002Natur.420..860C. doi:10.1038/nature01322. PMC 2803035. PMID 12490959.
- Gunn, L; Ding, C; Liu, M; Ma, Y; Qi, C; Cai, Y; Hu, X; Aggarwal, D; Zhang, HG; Yan, J (15 September 2012). "Opposing roles for complement component C5a in tumor progression and the tumor microenvironment". Journal of Immunology. 189 (6): 2985–94. doi:10.4049/jimmunol.1200846. PMC 3436956. PMID 22914051.
- Copland, JA; Sheffield-Moore, M; Koldzic-Zivanovic, N; Gentry, S; Lamprou, G; Tzortzatou-Stathopoulou, F; Zoumpourlis, V; Urban, RJ; Vlahopoulos, SA (June 2009). "Sex steroid receptors in skeletal differentiation and epithelial neoplasia: is tissue-specific intervention possible?". BioEssays. 31 (6): 629–41. doi:10.1002/bies.200800138. PMID 19382224.
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A; Allavena; Sica; Garlanda; Mantovani (July 2009). "Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability". Carcinogenesis (review). 30 (7): 1073–81. doi:10.1093/carcin/bgp127. PMID 19468060.CS1 maint: multiple names: authors list (link)
- Coussens LM, Werb Z (2002). "Inflammation and cancer". Nature. 420 (6917): 860–7. Bibcode:2002Natur.420..860C. doi:10.1038/nature01322. PMC 2803035. PMID 12490959.
- Chiba T, Marusawa H, Ushijima T (2012). "Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation" (PDF). Gastroenterology. 143 (3): 550–563. doi:10.1053/j.gastro.2012.07.009. hdl:2433/160134. PMID 22796521.
- Kawanishi S, Ohnishi S, Ma N, Hiraku Y, Murata M (2017). "Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis". Int J Mol Sci. 18 (8): 1808. doi:10.3390/ijms18081808. PMC 5578195. PMID 28825631.
- Ding, N., Maiuri, A. R., & O'Hagan, H. M. (2017). The emerging role of epigenetic modifiers in repair of DNA damage associated with chronic inflammatory diseases. Mutation Research/Reviews in Mutation Research. doi:10.1016/j.mrrev.2017.09.005
- Mantovani A, Allavena P, Sica A, Balkwill F (2008). "Cancer-related inflammation" (PDF). Nature. 454 (7203): 436–44. Bibcode:2008Natur.454..436M. doi:10.1038/nature07205. hdl:2434/145688. PMID 18650914.
- Larsen GL, Henson PM (1983). "Mediators of inflammation". Annu. Rev. Immunol. 1: 335–59. doi:10.1146/annurev.iy.01.040183.002003. PMID 6399978.
- Shacter E, Weitzman SA (2002). "Chronic inflammation and cancer". Oncology (Williston Park, N.Y.). 16 (2): 217–26, 229, discussion 230–2. PMID 11866137.
- Yu Y, Cui Y, Niedernhofer LJ, Wang Y (2016). "Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage". Chem. Res. Toxicol. 29 (12): 2008–2039. doi:10.1021/acs.chemrestox.6b00265. PMC 5614522. PMID 27989142.
- Dizdaroglu M (2012). "Oxidatively induced DNA damage: mechanisms, repair and disease". Cancer Lett. 327 (1–2): 26–47. doi:10.1016/j.canlet.2012.01.016. PMID 22293091.
- Nishida N, Kudo M (2013). "Oxidative stress and epigenetic instability in human hepatocarcinogenesis". Dig Dis. 31 (5–6): 447–53. doi:10.1159/000355243. PMID 24281019.
- Kawanishi S, Ohnishi S, Ma N, Hiraku Y, Oikawa S, Murata M (2016). "Nitrative and oxidative DNA damage in infection-related carcinogenesis in relation to cancer stem cells". Genes Environ. 38: 26. doi:10.1186/s41021-016-0055-7. PMC 5203929. PMID 28050219.
- O'Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, Casero RA, Sears CL, Baylin SB (2011). "Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands". Cancer Cell. 20 (5): 606–19. doi:10.1016/j.ccr.2011.09.012. PMC 3220885. PMID 22094255.
- Maiuri AR, Peng M, Sriramkumar S, Kamplain CM, DeStefano Shields CE, Sears CL, O'Hagan HM (2017). "Mismatch Repair Proteins Initiate Epigenetic Alterations during Inflammation-Driven Tumorigenesis". Cancer Res. 77 (13): 3467–3478. doi:10.1158/0008-5472.CAN-17-0056. PMC 5516887. PMID 28522752.
- Yamashita S, Kishino T, Takahashi T, Shimazu T, Charvat H, Kakugawa Y, Nakajima T, Lee YC, Iida N, Maeda M, Hattori N, Takeshima H, Nagano R, Oda I, Tsugane S, Wu MS, Ushijima T (2018). "Genetic and epigenetic alterations in normal tissues have differential impacts on cancer risk among tissues". Proc. Natl. Acad. Sci. U.S.A. 115 (6): 1328–1333. doi:10.1073/pnas.1717340115. PMC 5819434. PMID 29358395.
- Raza Y, Khan A, Farooqui A, Mubarak M, Facista A, Akhtar SS, Khan S, Kazi JI, Bernstein C, Kazmi SU (2014). "Oxidative DNA damage as a potential early biomarker of Helicobacter pylori associated carcinogenesis". Pathol. Oncol. Res. 20 (4): 839–46. doi:10.1007/s12253-014-9762-1. PMID 24664859.
- Deeks, Steven G. (1 January 2011). "HIV infection, inflammation, immunosenescence, and aging". Annual Review of Medicine. 62: 141–155. doi:10.1146/annurev-med-042909-093756. PMC 3759035. PMID 21090961.
- Klatt, Nichole R.; Chomont, Nicolas; Douek, Daniel C.; Deeks, Steven G. (1 July 2013). "Immune activation and HIV persistence: implications for curative approaches to HIV infection". Immunological Reviews. 254 (1): 326–342. doi:10.1111/imr.12065. PMC 3694608. PMID 23772629.
- Salazar-Gonzalez, J. F.; Martinez-Maza, O.; Nishanian, P.; Aziz, N.; Shen, L. P.; Grosser, S.; Taylor, J.; Detels, R.; Fahey, J. L. (1 August 1998). "Increased immune activation precedes the inflection point of CD4 T cells and the increased serum virus load in human immunodeficiency virus infection". The Journal of Infectious Diseases. 178 (2): 423–430. doi:10.1086/515629. PMID 9697722.
- Ipp, Hayley; Zemlin, Annalise (1 February 2013). "The paradox of the immune response in HIV infection: when inflammation becomes harmful". Clinica Chimica Acta; International Journal of Clinical Chemistry. 416: 96–99. doi:10.1016/j.cca.2012.11.025. PMID 23228847.
- Nasi, Milena; Pinti, Marcello; Mussini, Cristina; Cossarizza, Andrea (1 October 2014). "Persistent inflammation in HIV infection: established concepts, new perspectives". Immunology Letters. 161 (2): 184–8. doi:10.1016/j.imlet.2014.01.008. PMID 24487059.
- Milush, Jeffrey M.; Mir, Kiran D.; Sundaravaradan, Vasudha; Gordon, Shari N.; Engram, Jessica; Cano, Christopher A.; Reeves, Jacqueline D.; Anton, Elizabeth; O'Neill, Eduardo (1 March 2011). "Lack of clinical AIDS in SIV-infected sooty mangabeys with significant CD4+ T cell loss is associated with double-negative T cells". The Journal of Clinical Investigation. 121 (3): 1102–10. doi:10.1172/JCI44876. PMC 3049370. PMID 21317533.
- Rey-Cuillé, M. A.; Berthier, J. L.; Bomsel-Demontoy, M. C.; Chaduc, Y.; Montagnier, L.; Hovanessian, A. G.; Chakrabarti, L. A. (1 May 1998). "Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease". Journal of Virology. 72 (5): 3872–86. PMC 109612. PMID 9557672.
- Chahroudi, Ann; Bosinger, Steven E.; Vanderford, Thomas H.; Paiardini, Mirko; Silvestri, Guido (9 March 2012). "Natural SIV hosts: showing AIDS the door". Science. 335 (6073): 1188–93. Bibcode:2012Sci...335.1188C. doi:10.1126/science.1217550. PMC 3822437. PMID 22403383.
- Doitsh, Gilad; Galloway, Nicole L. K.; Geng, Xin; Yang, Zhiyuan; Monroe, Kathryn M.; Zepeda, Orlando; Hunt, Peter W.; Hatano, Hiroyu; Sowinski, Stefanie (23 January 2014). "Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection". Nature. 505 (7484): 509–514. Bibcode:2014Natur.505..509D. doi:10.1038/nature12940. PMC 4047036. PMID 24356306.
- Monroe, Kathryn M.; Yang, Zhiyuan; Johnson, Jeffrey R.; Geng, Xin; Doitsh, Gilad; Krogan, Nevan J.; Greene, Warner C. (24 January 2014). "IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV". Science. 343 (6169): 428–432. Bibcode:2014Sci...343..428M. doi:10.1126/science.1243640. PMC 3976200. PMID 24356113.
- Galloway, Nicole L. K.; Doitsh, Gilad; Monroe, Kathryn M.; Yang, Zhiyuan; Muñoz-Arias, Isa; Levy, David N.; Greene, Warner C. (8 September 2015). "Cell-to-Cell Transmission of HIV-1 Is Required to Trigger Pyroptotic Death of Lymphoid-Tissue-Derived CD4 T Cells". Cell Reports. 12 (10): 1555–63. doi:10.1016/j.celrep.2015.08.011. PMC 4565731. PMID 26321639.
- Doitsh, Gilad; Greene, Warner C. (9 March 2016). "Dissecting How CD4 T Cells Are Lost During HIV Infection". Cell Host & Microbe. 19 (3): 280–291. doi:10.1016/j.chom.2016.02.012. PMC 4835240. PMID 26962940.
- "Study of VX-765 in Subjects With Treatment-resistant Partial Epilepsy - Full Text View - ClinicalTrials.gov". clinicaltrials.gov. Retrieved 21 May 2016.
- Eming, S. A.; Krieg, T.; Davidson, J. M. (2007). "Inflammation in wound repair: molecular and cellular mechanisms". Journal of Investigative Dermatology. 127 (3): 514–525. doi:10.1038/sj.jid.5700701. PMID 17299434.
- Ashcroft, G. S.; Yang, X; Glick, A. B.; Weinstein, M; Letterio, J. L.; Mizel, D. E.; Anzano, M; Greenwell-Wild, T; Wahl, S. M.; Deng, C; Roberts, A. B. (1999). "Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response". Nat Cell Biol. 1 (5): 260–6. doi:10.1038/12971. PMID 10559937.
- Ashcroft, G. S. (1999). "Bidirectional regulation of macrophage function by TGF-β". Microbes Infect. 1 (15): 1275–82. doi:10.1016/S1286-4579(99)00257-9. PMID 10611755.
- Werner, F; Jain, M. K.; Feinberg, M. W.; Sibinga, N. E.; Pellacani, A; Wiesel, P; Chin, M. T.; Topper, J. N.; Perrella, M. A.; Lee, M. E. (2000). "Transforming growth factor-β1 inhibition of macrophage activation is mediated via Smad3". J Biol Chem. 275 (47): 36653–8. doi:10.1074/jbc.M004536200. PMID 10973958.
- Sato, Y.; Ohshima, T.; Kondo, T. (1999). "Regulatory role of endogenous interleukin-10 in cutaneous inflammatory response of murine wound healing". Biochem Biophys Res Commun. 265 (1): 194–9. doi:10.1006/bbrc.1999.1455. PMID 10548513.
- Serhan, C. N. (2008). "Controlling the resolution of acute inflammation: a new genus of dual anti-inflammatory and proresolving mediators". J Periodontol. 79 (8 Suppl): 1520–6. doi:10.1902/jop.2008.080231. PMID 18673006.
- Headland SE, Norling LV (2015). "The resolution of inflammation: Principles and challenges". Seminars in Immunology. 27 (3): 149–60. doi:10.1016/j.smim.2015.03.014. PMID 25911383.
- Greenhalgh, D. G. (1998). "The role of apoptosis in wound healing". Int J Biochem Cell Biol. 30 (9): 1019–30. doi:10.1016/S1357-2725(98)00058-2. PMID 9785465.
- Jiang, D; Liang, J; Fan, J; Yu, S; Chen, S; Luo, Y; Prestwich, G. D.; Mascarenhas, M. M.; Garg, H. G.; Quinn, D. A.; Homer, R. J.; Goldstein, D. R.; Bucala, R; Lee, P. J.; Medzhitov, R; Noble, P. W. (2005). "Regulation of lung injury and repair by Toll-like receptors and hyaluronan". Nat Med. 11 (11): 1173–9. doi:10.1038/nm1315. PMID 16244651.
- Teder, P. (2002). "Resolution of lung inflammation by CD44". Science. 296 (5565): 155–8. Bibcode:2002Sci...296..155T. doi:10.1126/science.1069659. PMID 11935029.
- McQuibban, G. A.; Gong, J. H.; Tam, E. M.; McCulloch, C. A.; Clark-Lewis, I; Overall, C. M. (2000). "Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3". Science. 289 (5482): 1202–6. Bibcode:2000Sci...289.1202M. doi:10.1126/science.289.5482.1202. PMID 10947989.
- Serhan CN, Savill J; Savill (2005). "Resolution of inflammation: the beginning programs the end". Nat. Immunol. 6 (12): 1191–7. doi:10.1038/ni1276. PMID 16369558.
- Berk, M; Williams, L. J.; Jacka, F. N.; O'Neil, A; Pasco, J. A.; Moylan, S; Allen, N. B.; Stuart, A. L.; Hayley, A. C.; Byrne, M. L.; Maes, M (2013). "So depression is an inflammatory disease, but where does the inflammation come from?". BMC Medicine. 11: 200. doi:10.1186/1741-7015-11-200. PMC 3846682. PMID 24228900.
- Cox, William T. L.; Abramson, Lyn Y.; Devine, Patricia G.; Hollon, Steven D. (2012). "Stereotypes, Prejudice, and Depression: The Integrated Perspective". Perspectives on Psychological Science. 7 (5): 427–449. doi:10.1177/1745691612455204. PMID 26168502.
- Kiecolt-Glaser, Janice K.; Derry, Heather M.; Fagundes, Christopher P. (November 2015). "Inflammation: Depression Fans the Flames and Feasts on the Heat". American Journal of Psychiatry. 172 (11): 1075–91. doi:10.1176/appi.ajp.2015.15020152. PMC 6511978. PMID 26357876.
- Williams, Caroline (4 January 2015). "Is depression a kind of allergic reaction?". the Guardian.
- Brietzke, Elisa; Stertz, Laura; Fernandes, Brisa Simões; Kauer-Sant'Anna, Marcia; Mascarenhas, Marcello; Escosteguy Vargas, Andréia; Chies, José Artur; Kapczinski, Flávio (2009). "Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder". Journal of Affective Disorders. 116 (3): 214–7. doi:10.1016/j.jad.2008.12.001. PMID 19251324.
- Müller, N; Schwarz, M J; Dehning, S; Douhe, A; Cerovecki, A; Goldstein-Müller, B; Spellmann, I; Hetzel, G; Maino, K; Kleindienst, N; Möller, H-J; Arolt, V; Riedel, M (2006). "The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: Results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine". Molecular Psychiatry. 11 (7): 680–4. doi:10.1038/sj.mp.4001805. PMID 16491133.
- Canli, Turhan (2014). "Reconceptualizing major depressive disorder as an infectious disease". Biology of Mood & Anxiety Disorders. 4: 10. doi:10.1186/2045-5380-4-10. PMC 4215336. PMID 25364500.
- Parimisetty A, Dorsemans AC, Awada R, Ravanan P, Diotel N, Lefebvre d'Hellencourt C (24 March 2016). "Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research". J Neuroinflammation (Review). 13 (1): 67. doi:10.1186/s12974-016-0530-x. PMC 4806498. PMID 27012931.
- Kershaw, E. E.; Flier, J. S. (2004). "Adipose tissue as an endocrine organ". J Clin Endocrinol Metab. 89 (6): 2548–56. doi:10.1210/jc.2004-0395. PMID 15181022.
- Bastard J, et al. (2000). "Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss". J Clin Endocrinol Metab. 85 (9): 3338–42. doi:10.1210/jc.85.9.3338. PMID 10999830.
- Mohamed-Ali V, et al. (2001). "beta-Adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies". J Clin Endocrinol Metab. 86 (12): 5864–9. doi:10.1210/jc.86.12.5864. PMID 11739453.
- Loffreda, S; Yang, S. Q.; Lin, H. Z.; Karp, C. L.; Brengman, M. L.; Wang, D. J.; Klein, A. S.; Bulkley, G. B.; Bao, C; Noble, P. W.; Lane, M. D.; Diehl, A. M. (1998). "Leptin regulates proinflammatory immune responses". FASEB J. 12 (1): 57–65. doi:10.1096/fasebj.12.1.57. PMID 9438411.
- Esposito, K; Nappo, F; Marfella, R; Giugliano, G; Giugliano, F; Ciotola, M; Quagliaro, L; Ceriello, A; Giugliano, D (2002). "Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress". Circulation. 106 (16): 2067–72. doi:10.1161/01.CIR.0000034509.14906.AE. PMID 12379575.
- Petersen, A. M.; Pedersen, B. K. (2005). "The anti-inflammatory effect of exercise". J Appl Physiol. 98 (4): 1154–62. doi:10.1152/japplphysiol.00164.2004. PMID 15772055.
- Rogowski, O; Shapira, I; Bassat, O. K.; Chundadze, T; Finn, T; Berliner, S; Steinvil, A (2010). "Waist circumference as the predominant contributor to the micro-inflammatory response in the metabolic syndrome: a cross sectional study". Journal of Inflammation. 7: 35. doi:10.1186/1476-9255-7-35. PMC 2919526. PMID 20659330.
- Goldfine, Allison B.; Shoelson, Steven E. (3 January 2017). "Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk". Journal of Clinical Investigation. 127 (1): 83–93. doi:10.1172/jci88884. ISSN 0021-9738. PMC 5199685. PMID 28045401.
- Choi, J.; Joseph, L.; Pilote, L. (2013). "Obesity and C-reactive protein in various populations: a systematic review and meta-analysis". Obesity Reviews. 14 (3): 232–244. doi:10.1111/obr.12003. ISSN 1467-7881. PMID 23171381.
- Wilmore, Jack (2008). Physiology of Sport and Exercise. Champaign, IL: Human Kinetics. pp. 26–36, 98–120, 186–250, 213–218. ISBN 978-0-7360-5583-3.
- "Physiology of Sport and Exercise 5E: Delayed-Onset Muscle Soreness". human-kinetics. Retrieved 4 June 2017.
- Toth, M. J.; Matthews, DE; Tracy, RP; Previs, MJ (29 December 2004). "Age-related differences in skeletal muscle protein synthesis: relation to markers of immune activation". AJP: Endocrinology and Metabolism. 288 (5): E883–E891. doi:10.1152/ajpendo.00353.2004. PMID 15613683.
- Mikkelsen, U. R.; Langberg, H.; Helmark, I. C.; Skovgaard, D.; Andersen, L. L.; Kjaer, M.; Mackey, A. L. (27 August 2009). "Local NSAID infusion inhibits satellite cell proliferation in human skeletal muscle after eccentric exercise". Journal of Applied Physiology. 107 (5): 1600–11. doi:10.1152/japplphysiol.00707.2009. PMC 3774508. PMID 19713429.
- Trappe, TA; White, F; Lambert, CP; Cesar, D; Hellerstein, M; Evans, WJ (March 2002). "Effect of ibuprofen and acetaminophen on postexercise muscle protein synthesis". American Journal of Physiology. Endocrinology and Metabolism. 282 (3): E551–6. doi:10.1152/ajpendo.00352.2001. PMID 11832356.
- Takagi, Ryo; Fujita, Naoto; Arakawa, Takamitsu; Kawada, Shigeo; Ishii, Naokata; Miki, Akinori (1 February 2011). "Influence of icing on muscle regeneration after crush injury to skeletal muscles in rats". Journal of Applied Physiology. 110 (2): 382–8. doi:10.1152/japplphysiol.01187.2010. PMID 21164157.
- Mikkelsen, U. R.; Langberg, H.; Helmark, I. C.; Skovgaard, D.; Andersen, L. L.; Kjær, M.; Mackey, A. L. (1 November 2009). "Local NSAID infusion inhibits satellite cell proliferation in human skeletal muscle after eccentric exercise". Journal of Applied Physiology. 107 (5): 1600–11. doi:10.1152/japplphysiol.00707.2009. PMC 3774508. PMID 19713429.
- Roberts, Llion A.; Raastad, Truls; Markworth, James F.; Figueiredo, Vandre C.; Egner, Ingrid M.; Shield, Anthony; Cameron-Smith, David; Coombes, Jeff S.; Peake, Jonathan M. (15 September 2015). "Post-exercise cold water immersion attenuates acute anabolic signalling and long-term adaptations in muscle to strength training". The Journal of Physiology. 593 (18): 4285–4301. doi:10.1113/JP270570. PMC 4594298. PMID 26174323.
- Marimuthu, K.; Murton, A. J.; Greenhaff, P. L. (28 October 2010). "Mechanisms regulating muscle mass during disuse atrophy and rehabilitation in humans". Journal of Applied Physiology. 110 (2): 555–560. doi:10.1152/japplphysiol.00962.2010. PMID 21030670.
- Cannon, Joseph G.; St. Pierre, Barbara A. (1 January 1998). "Cytokines in exertion-induced skeletal muscle injury". Molecular and Cellular Biochemistry. 179 (1/2): 159–168. doi:10.1023/A:1006828425418. PMID 9543358.
- Lang, Charles H.; Hong-Brown, Ly; Frost, Robert A. (10 November 2004). "Cytokine inhibition of JAK-STAT signaling: a new mechanism of growth hormone resistance". Pediatric Nephrology. 20 (3): 306–312. doi:10.1007/s00467-004-1607-9. PMID 15549417.
- Pedersen, BK; Toft, AD (August 2000). "Effects of exercise on lymphocytes and cytokines". British Journal of Sports Medicine. 34 (4): 246–51. doi:10.1136/bjsm.34.4.246. PMC 1724218. PMID 10953894.
- Bruunsgaard, H; Galbo, H; Halkjaer-Kristensen, J; Johansen, TL; MacLean, DA; Pedersen, BK (15 March 1997). "Exercise-induced increase in serum interleukin-6 in humans is related to muscle damage". The Journal of Physiology. 499 (Pt 3): 833–41. doi:10.1113/jphysiol.1997.sp021972. PMC 1159298. PMID 9130176.
- McKay, Bryon R.; De Lisio, Michael; Johnston, Adam P. W.; O'Reilly, Ciara E.; Phillips, Stuart M.; Tarnopolsky, Mark A.; Parise, Gianni; Hotchin, Neil; Johnston, Adam P. W.; O'Reilly, Ciara E.; Phillips, Stuart M.; Tarnopolsky, Mark A.; Parise, Gianni (23 June 2009). Hotchin, Neil (ed.). "Association of Interleukin-6 Signalling with the Muscle Stem Cell Response Following Muscle-Lengthening Contractions in Humans". PLoS ONE. 4 (6): e6027. Bibcode:2009PLoSO...4.6027M. doi:10.1371/journal.pone.0006027. PMC 2696599. PMID 19554087.CS1 maint: multiple names: authors list (link)
- MacIntyre, Donna L.; Sorichter, Stephan; Mair, Johannes; McKenzie, Donald C.; Berg, Aloys (11 March 2001). "Markers of inflammation and myofibrillar proteins following eccentric exercise in humans". European Journal of Applied Physiology. 84 (3): 180–6. doi:10.1007/s004210170002. PMID 11320633.
- Louis, E; Raue, U; Yang, Y; Jemiolo, B; Trappe, S (November 2007). "Time course of proteolytic, cytokine, and myostatin gene expression after acute exercise in human skeletal muscle". Journal of Applied Physiology. 103 (5): 1744–51. doi:10.1152/japplphysiol.00679.2007. PMID 17823296.
- Serrano, AL; Baeza-Raja, B; Perdiguero, E; Jardí, M; Muñoz-Cánoves, P (January 2008). "Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy". Cell Metabolism. 7 (1): 33–44. doi:10.1016/j.cmet.2007.11.011. PMID 18177723.
- Grounds, MD; White, JD; Rosenthal, N; Bogoyevitch, MA (May 2002). "The role of stem cells in skeletal and cardiac muscle repair". Journal of Histochemistry and Cytochemistry. 50 (5): 589–610. doi:10.1177/002215540205000501. PMID 11967271.
- Hawke, TJ; Garry, DJ (August 2001). "Myogenic satellite cells: physiology to molecular biology". Journal of Applied Physiology. 91 (2): 534–51. doi:10.1152/jappl.2001.91.2.534. PMID 11457764.
- Hawke, TJ (April 2005). "Muscle stem cells and exercise training". Exercise and Sport Sciences Reviews. 33 (2): 63–8. doi:10.1097/00003677-200504000-00002. PMID 15821426.
- Kadi, F; Eriksson, A; Holmner, S; Butler-Browne, GS; Thornell, LE (March 1999). "Cellular adaptation of the trapezius muscle in strength-trained athletes". Histochemistry and Cell Biology. 111 (3): 189–95. doi:10.1007/s004180050348. PMID 10094415.
- Eriksson, A; Kadi, F; Malm, C; Thornell, LE (August 2005). "Skeletal muscle morphology in power-lifters with and without anabolic steroids". Histochemistry and Cell Biology. 124 (2): 167–75. doi:10.1007/s00418-005-0029-5. PMID 16059740.
- Mikkelsen, UR; Schjerling, P; Helmark, IC; Reitelseder, S; Holm, L; Skovgaard, D; Langberg, H; Kjaer, M; Heinemeier, KM (October 2011). "Local NSAID infusion does not affect protein synthesis and gene expression in human muscle after eccentric exercise". Scandinavian Journal of Medicine & Science in Sports. 21 (5): 630–44. doi:10.1111/j.1600-0838.2010.01170.x. PMID 20738823.
- Pedersen, BK (July 2013). Muscle as a secretory organ. Comprehensive Physiology. 3. pp. 1337–62. doi:10.1002/cphy.c120033. ISBN 9780470650714. PMID 23897689.
- Visser, M; Pahor, M; Taaffe, DR; Goodpaster, BH; Simonsick, EM; Newman, AB; Nevitt, M; Harris, TB (May 2002). "Relationship of interleukin-6 and tumor necrosis factor-alpha with muscle mass and muscle strength in elderly men and women: the Health ABC Study". The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 57 (5): M326–32. doi:10.1093/gerona/57.5.M326. PMID 11983728.
- Reardon, KA; Davis, J; Kapsa, RM; Choong, P; Byrne, E (July 2001). "Myostatin, insulin-like growth factor-1, and leukemia inhibitory factor mRNAs are upregulated in chronic human disuse muscle atrophy". Muscle & Nerve. 24 (7): 893–9. doi:10.1002/mus.1086. PMID 11410916.
- Shih, Michael. "Skeletal Muscle Hypertrophy Is Regulated via AKT/mTOR Pathway." BioCarta. Web. 21 March 2011. "Blogger". Archived from the original on 14 September 2010. Retrieved 2011-03-21.
- Lang, CH; Frost, RA (January 2007). "Sepsis-induced suppression of skeletal muscle translation initiation mediated by tumor necrosis factor alpha". Metabolism: Clinical and Experimental. 56 (1): 49–57. doi:10.1016/j.metabol.2006.08.025. PMID 17161226.
- García-Martínez, C; López-Soriano, FJ; Argilés, JM (11 August 1993). "Acute treatment with tumour necrosis factor-alpha induces changes in protein metabolism in rat skeletal muscle". Molecular and Cellular Biochemistry. 125 (1): 11–8. doi:10.1007/BF00926829. PMID 8264567.
- Janssen, SP; Gayan-Ramirez, G; Van den Bergh, A; Herijgers, P; Maes, K; Verbeken, E; Decramer, M (1 March 2005). "Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats". Circulation. 111 (8): 996–1005. doi:10.1161/01.CIR.0000156469.96135.0D. PMID 15710765.CS1 maint: multiple names: authors list (link)
- Lang, CH; Frost, RA; Vary, TC (August 2007). "Regulation of muscle protein synthesis during sepsis and inflammation". American Journal of Physiology. Endocrinology and Metabolism. 293 (2): E453–9. doi:10.1152/ajpendo.00204.2007. PMID 17505052.
- Jurasinski, CV; Vary, TC (November 1995). "Insulin-like growth factor I accelerates protein synthesis in skeletal muscle during sepsis". The American Journal of Physiology. 269 (5 Pt 1): E977–81. doi:10.1152/ajpendo.1995.269.5.E977. PMID 7491951.
- Vary, TC; Kimball, SR (February 1992). "Regulation of hepatic protein synthesis in chronic inflammation and sepsis". The American Journal of Physiology. 262 (2 Pt 1): C445–52. doi:10.1152/ajpcell.1992.262.2.C445. PMID 1371643.
- Lang, CH; Frost, RA; Bronson, SK; Lynch, CJ; Vary, TC (June 2010). "Skeletal muscle protein balance in mTOR heterozygous mice in response to inflammation and leucine". American Journal of Physiology. Endocrinology and Metabolism. 298 (6): E1283–94. doi:10.1152/ajpendo.00676.2009. PMC 2886531. PMID 20388826.
- Smith JK; Dykes R; Douglas JE; Krishnaswamy G; Berk S (12 May 1999). "Long-term exercise and atherogenic activity of blood mononuclear cells in persons at risk of developing ischemic heart disease". JAMA: the Journal of the American Medical Association. 281 (18): 1722–7. doi:10.1001/jama.281.18.1722. PMID 10328073.
- McFarlin, BK; Flynn, MG; Phillips, MD; Stewart, LK; Timmerman, KL (October 2005). "Chronic resistance exercise training improves natural killer cell activity in older women". The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 60 (10): 1315–8. doi:10.1093/gerona/60.10.1315. PMID 16282566.
- Stewart LK; Flynn MG; Campbell WW; Craig BA; Robinson JP; McFarlin, BK; Timmerman KL; Coen PM; Felker J; Talbert E (September 2005). "Influence of exercise training and age on CD14+ cell-surface expression of toll-like receptor 2 and 4". Brain, Behavior, and Immunity. 19 (5): 389–97. doi:10.1016/j.bbi.2005.04.003. PMID 15963685.
- Gleeson, M (November 2006). "Immune system adaptation in elite athletes". Current Opinion in Clinical Nutrition and Metabolic Care. 9 (6): 659–65. doi:10.1097/01.mco.0000247476.02650.18. PMID 17053416.
- Pedersen, BK; Hoffman-Goetz, L (July 2000). "Exercise and the immune system: regulation, integration, and adaptation". Physiological Reviews. 80 (3): 1055–81. doi:10.1152/physrev.2000.80.3.1055. PMID 10893431.
- Ploeger, H. E.; Takken, T; De Greef, M. H.; Timmons, B. W. (2009). "The effects of acute and chronic exercise on inflammatory markers in children and adults with a chronic inflammatory disease: a systematic review". Exercise Immunology Review. 15: 6–41. PMID 19957870.
- Nicklas BJ; Hsu FC; Brinkley TJ; Church T; Goodpaster BH; Kritchevsky SB; Pahor, M (November 2008). "Exercise training and plasma C-reactive protein and interleukin-6 in elderly people". Journal of the American Geriatrics Society. 56 (11): 2045–52. doi:10.1111/j.1532-5415.2008.01994.x. PMC 2683336. PMID 19016938.
- Timmerman KL; Flynn MG; Coen PM; Markofski MM; Pence BD (November 2008). "Exercise training-induced lowering of inflammatory (CD14+CD16+) monocytes: a role in the anti-inflammatory influence of exercise?". Journal of Leukocyte Biology. 84 (5): 1271–8. doi:10.1189/jlb.0408244. PMID 18664531.
- Mackinnon LT (July 2000). "Chronic exercise training effects on immune function". Med Sci Sports Exerc. 32 (7 Suppl): S369–76. doi:10.1097/00005768-200007001-00001. PMID 10910293.
- Suzuki Katsuhiko; Nakaji Shigeyuki; Yamada Mutsuo; Liu Qiang; Kurakake Shigeyoshi; Okamura Noriyoshi; Kumae Takashi; Umeda Takashi; Sugawara Kazuo (February 2003). "Impact of a competitive marathon race on systemic cytokine and neutrophil responses". Medicine & Science in Sports & Exercise. 35 (2): 348–55. doi:10.1249/01.MSS.0000048861.57899.04. PMID 12569227.
- Bruunsgaard H, Galbo H, Halkjaer-Kristensen J, Johansen TL, MacLean DA, Pedersen BK; Galbo; Halkjaer-Kristensen; Johansen; MacLean; Pedersen (March 1997). "Exercise-induced increase in serum interleukin-6 in humans is related to muscle damage". Journal of Physiology. 499 (Pt 3): 833–41. doi:10.1113/jphysiol.1997.sp021972. PMC 1159298. PMID 9130176.CS1 maint: multiple names: authors list (link)
- Pedersen BK (July 2013). Muscle as a secretory organ. Compr Physiol. 3. pp. 1337–62. doi:10.1002/cphy.c120033. ISBN 9780470650714. PMID 23897689.
- Brandt C; Pedersen BK (2010). "The role of exercise-induced myokines in muscle homeostasis and the defense against chronic diseases". Journal of Biomedicine & Biotechnology. 2010: 1–6. doi:10.1155/2010/520258. PMC 2836182. PMID 20224659.
- Pilon, Brad. "Inflammation Affects Your Ability to Build Muscle" Inflammation Theory
External links
| Wikimedia Commons has media related to Inflammations. |
- Inflammation at the US National Library of Medicine Medical Subject Headings (MeSH)