Renal tubular acidosis
Renal tubular acidosis (RTA) is a medical condition that involves an accumulation of acid in the body due to a failure of the kidneys to appropriately acidify the urine.[1] In renal physiology, when blood is filtered by the kidney, the filtrate passes through the tubules of the nephron, allowing for exchange of salts, acid equivalents, and other solutes before it drains into the bladder as urine. The metabolic acidosis that results from RTA may be caused either by failure to reabsorb sufficient bicarbonate ions (which are alkaline) from the filtrate in the early portion of the nephron (the proximal tubule) or by insufficient secretion of hydrogen ions (which are acidic) into the latter portions of the nephron (the distal tubule). Although a metabolic acidosis also occurs in those with chronic kidney disease, the term RTA is reserved for individuals with poor urinary acidification in otherwise well-functioning kidneys. Several different types of RTA exist, which all have different syndromes and different causes. RTA is usually an incidental finding based on routine blood draws that show abnormal results. Clinically, patients may present with vague symptoms such as dehydration, mental status changes, or delayed growth in adolescents. [2]
| Renal tubular acidosis | |
|---|---|
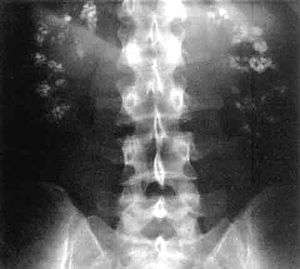 | |
| Significant bilateral nephrocalcinosis (calcification of the kidneys) on a frontal X-ray (radiopacities (white) in the right upper and left upper quadrant of the image), as seen in distal renal tubular acidosis | |
| Specialty | Nephrology |
The word acidosis refers to the tendency for RTA to cause an excess of acid, which lowers the blood's pH. When the blood pH is below normal (7.35), this is called acidemia. The metabolic acidosis caused by RTA is a normal anion gap acidosis.
Types
An overview of types 1, 2, and 4 is presented below (type 3 is usually excluded from modern classifications):
| Type | Type 1 | Type 2 | Type 4 |
|---|---|---|---|
| Location | Collecting Tubules, distal tubules | Proximal tubules | Adrenal |
| Acidemia | Yes (very severe) | Yes | Mild when present |
| Potassium | Hypokalemia | Hypokalemia | Hyperkalemia |
| Pathophysiology | Failure of α intercalated cells to secrete H+ and reclaim K+ | Failure of proximal tubular cells to reabsorb HCO− 3 |
Deficiency of aldosterone, or a resistance to its effects, (hypoaldosteronism or pseudohypoaldosteronism) |
Type 1: distal
Distal RTA (dRTA) is the classical form of RTA, being the first described. Distal RTA is characterized by a failure of H+ secretion into lumen of nephron by the alpha intercalated cells of the medullary collecting duct of the distal nephron.
This failure of acid secretion may be due to a number of causes, and it leads to an inability to acidify the urine to a pH of less than 5.3. Because renal excretion is the primary means of eliminating H+
from the body, there is consequently a tendency towards acidemia. There is an inability to excrete H+ while K+
cannot be reclaimed by the cell, leading to acidemia (as H+
builds up in the body) and hypokalemia (as K+
cannot be reabsorbed by the alpha cell).
This leads to the clinical features of dRTA;[1] In other words, the intercalated cells' apical H+/K+ antiporter is non-functional, resulting in proton retention and potassium excretion. Since calcium phosphate stones demonstrate a proclivity for deposition at higher pHs (alkaline), the substance of the kidney develops stones bilaterally; this does not occur in the other RTA types.
- Normal anion gap metabolic acidosis/acidemia
- Hypokalemia, Hypocalcemia, Hyperchloremia
- Urinary stone formation (related to alkaline urine, hypercalciuria, and low urinary citrate).[3]
- Nephrocalcinosis (deposition of calcium in the substance of the kidney)
- Bone demineralisation (causing rickets in children and osteomalacia in adults
- Growth deficiency
- Medullary cysts
- Sensorineural hearing loss
- Hereditary hemolytic anemia
Distal RTA has also been linked to specific genetic mutations that will alter when the disease will present in the patient’s life. Patient’s with mutations in ATP6V1B1 and ATP6V0A4 will present with symptoms within the first year of life, while those with mutation of the SLC4A1 have delayed onset around 10 years of age.[4] Electrolyte imbalances remain the same, while in severe cases symptoms can advance to amino aciduria and hyperammonemia. [5]
dRTA is the most common form of RTA diagnosed in Western countries, and can be classified as either hereditary (primary) or acquired (secondary). Primary RTA generally results from systemic and autoimmune diseases[6] or drug and toxin exposure in adults, whereas pediatric RTA results from genetic defects in the proteins that facilitate urine acidification at the distal tubule. Hereditary dRTA generally presents as failure to thrive during the first several months of life. Other common clinical manifestations in children include a variety of gastrointestinal and urinary symptoms, including polyuria, polydipsia, constipation, diarrhea, bouts of dehydration, and decreased appetite.[7]
Type 2: proximal
Proximal RTA (pRTA) is caused by a failure of the proximal tubular cells to reabsorb filtered bicarbonate from the urine, leading to urinary bicarbonate wasting and subsequent acidemia. Reabsorption of bicarbonate is typically 80-90% in the proximal tubule and failure of this process leads to decreased systemic buffer and metabolic acidosis.[8] The distal intercalated cells function normally, so the acidemia is less severe than dRTA and the alpha intercalated cells can produce H+ to acidify the urine to a pH of less than 5.3.[9] pRTA also has several causes, and may occasionally be present as a solitary defect, but is usually associated with a more generalized dysfunction of the proximal tubular cells called Fanconi syndrome, in which there is also phosphaturia, glycosuria, aminoaciduria, uricosuria, and tubular proteinuria.
The principal feature of Fanconi syndrome is bone demineralization (osteomalacia or rickets) due to phosphate wasting.
Type 3: combined proximal and distal
In some patients, RTA shares features of both dRTA and pRTA. This rare pattern was observed in the 1960s and 1970s as a transient phenomenon in infants and children with dRTA (possibly in relation with some exogenous factor such as high salt intake) and is no longer observed.[10] This form of RTA has also been referred to as juvenile RTA.[11]
Combined dRTA and pRTA is also observed as the result of inherited carbonic anhydrase II deficiency. Mutations in the gene encoding this enzyme give rise to an autosomal recessive syndrome of osteopetrosis, renal tubular acidosis, cerebral calcification, and mental retardation.[12][13][14] It is very rare and cases from all over the world have been reported, of which about 70% are from the Maghreb region of North Africa, possibly due to the high prevalence of consanguinity there.[15] The kidney problems are treated as described above. There is no treatment for the osteopetrosis or cerebral calcification.
Type 3 is rarely discussed.[16] Most comparisons of RTA are limited to a comparison of types 1, 2, and 4.
Type 4: absolute hypoaldosteronism or aldosterone insensitivity
Type 4 RTA is not actually a tubular disorder at all nor does it have a clinical syndrome similar to the other types of RTA described above. It was included in the classification of renal tubular acidoses as it is associated with a mild (normal anion gap) metabolic acidosis due to a physiological reduction in proximal tubular ammonium excretion (impaired ammoniagenesis), which is secondary to hypoaldosteronism, and results in a decrease in urine buffering capacity. Its cardinal feature is hyperkalemia, and measured urinary acidification is normal, hence it is often called hyperkalemic RTA or tubular hyperkalemia.[16]
Causes include:
- Aldosterone deficiency (hypoaldosteronism): Primary vs. hyporeninemic (including diabetic nephropathy)
- Aldosterone resistance
History
Renal tubular acidosis was first described in 1935 by Lightwood and 1936 by Butler et al. in children.[17][18] Baines et al. first described it in adults in 1945.[19]
Donald L. Lewis postulated the character Tiny Tim, of A Christmas Carol, was suffering from renal tubular acidosis.[20]
Researchers published PLOS ONE in 2009 speculated that the infamously afflicted Charles II of Spain may have suffered from renal tubular acidosis in tandem with combined pituitary hormone deficiency.[21]
See also
- Charles II of Spain, who is speculated to have suffered with dRTA
- Hyperchloremic acidosis
- Hypokalemic acidosis
References
- Laing CM, Toye AM, Capasso G, Unwin RJ (June 2005). "Renal tubular acidosis: developments in our understanding of the molecular basis". The International Journal of Biochemistry & Cell Biology. 37 (6): 1151–61. doi:10.1016/j.biocel.2005.01.002. PMID 15778079.
- Yaxley J, Pirrone C (2016-12-21). "Review of the Diagnostic Evaluation of Renal Tubular Acidosis". The Ochsner Journal. 16 (4): 525–530. PMC 5158160. PMID 27999512.
- Buckalew VM (March 1989). "Nephrolithiasis in renal tubular acidosis". The Journal of Urology. 141 (3 Pt 2): 731–7. doi:10.1016/S0022-5347(17)40997-9. PMID 2645431.
- Alonso-Varela M, Gil-Peña H, Coto E, Gómez J, Rodríguez J, Rodríguez-Rubio E, Santos F (September 2018). "Distal renal tubular acidosis. Clinical manifestations in patients with different underlying gene mutations". Pediatric Nephrology. 33 (9): 1523–1529. doi:10.1007/s00467-018-3965-8. hdl:10651/48680. PMID 29725771.
- Clericetti CM, Milani GP, Lava SA, Bianchetti MG, Simonetti GD, Giannini O (March 2018). "Hyperammonemia associated with distal renal tubular acidosis or urinary tract infection: a systematic review". Pediatric Nephrology. 33 (3): 485–491. doi:10.1007/s00467-017-3829-7. PMID 29134448.
- Santos F, Ordóñez FA, Claramunt-Taberner D, Gil-Peña H (December 2015). "Clinical and laboratory approaches in the diagnosis of renal tubular acidosis". Pediatric Nephrology. 30 (12): 2099–107. doi:10.1007/s00467-015-3083-9. PMID 25823989.
- Soleimani M, Rastegar A (September 2016). "Pathophysiology of Renal Tubular Acidosis: Core Curriculum 2016". American Journal of Kidney Diseases. 68 (3): 488–98. doi:10.1053/j.ajkd.2016.03.422. PMID 27188519.
- Pereira PC, Miranda DM, Oliveira EA, Silva AC (March 2009). "Molecular pathophysiology of renal tubular acidosis". Current Genomics. 10 (1): 51–9. doi:10.2174/138920209787581262. PMC 2699831. PMID 19721811.
- Rodriguez Soriano J, Boichis H, Stark H, Edelmann CM (March 1967). "Proximal renal tubular acidosis. A defect in bicarbonate reabsorption with normal urinary acidification". Pediatric Research. 1 (2): 81–98. doi:10.1203/00006450-196703000-00001. PMID 6029811.
- Rodríguez Soriano J (August 2002). "Renal tubular acidosis: the clinical entity". Journal of the American Society of Nephrology. 13 (8): 2160–70. doi:10.1097/01.ASN.0000023430.92674.E5. PMID 12138150.
- de Jong PE, Koomans HA, Weening JJ (2000). Klinische Nefrologie (3rd ed.). Maarssen: Elsevier. pp. 141–2.
- Sly WS, Whyte MP, Sundaram V, Tashian RE, Hewett-Emmett D, Guibaud P, et al. (July 1985). "Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification". The New England Journal of Medicine. 313 (3): 139–45. doi:10.1056/NEJM198507183130302. PMID 3925334.
- Shah GN, Bonapace G, Hu PY, Strisciuglio P, Sly WS (September 2004). "Carbonic anhydrase II deficiency syndrome (osteopetrosis with renal tubular acidosis and brain calcification): novel mutations in CA2 identified by direct sequencing expand the opportunity for genotype-phenotype correlation". Human Mutation. 24 (3): 272. doi:10.1002/humu.9266. PMID 15300855.
- Pushkin A, Abuladze N, Gross E, Newman D, Tatishchev S, Lee I, et al. (August 2004). "Molecular mechanism of kNBC1-carbonic anhydrase II interaction in proximal tubule cells". The Journal of Physiology. 559 (Pt 1): 55–65. doi:10.1113/jphysiol.2004.065110. PMC 1665076. PMID 15218065.
- Fathallah DM, Bejaoui M, Lepaslier D, Chater K, Sly WS, Dellagi K (May 1997). "Carbonic anhydrase II (CA II) deficiency in Maghrebian patients: evidence for founder effect and genomic recombination at the CA II locus". Human Genetics. 99 (5): 634–7. doi:10.1007/s004390050419. PMID 9150731.
- Hyporeninemic Hypoaldosteronism at eMedicine
- Lightwood R. (1935). "Communication no. 1". Arch Dis Child. 10 (57): 205–6. doi:10.1136/adc.10.57.205. PMC 1975385.
- Butler AM, Wilson JL, Farber S (1936). "Dehydration and acidosis with calcification at renal tubules". The Journal of Pediatrics. 8 (4): 489–99. doi:10.1016/s0022-3476(36)80111-5.
- Baines AM, Barelay JA, Cooke WT (1945). "Nephrocalcinosis associated with hyperchloremia and low plasma-bicarbonate". Q J Med. 14: 113–23.
- Lewis DW (December 1992). "What was wrong with Tiny Tim?". American Journal of Diseases of Children. 146 (12): 1403–7. doi:10.1001/archpedi.1992.02160240013002. PMID 1340779.
- Alvarez G, Ceballos FC, Quinteiro C (15 April 2009). "The role of inbreeding in the extinction of a European royal dynasty". PLOS ONE. 4 (4): e5174. Bibcode:2009PLoSO...4.5174A. doi:10.1371/journal.pone.0005174. PMC 2664480. PMID 19367331.
External links
| Classification | |
|---|---|
| External resources |