Bartter syndrome
Bartter syndrome is a rare inherited disease characterised by a defect in the thick ascending limb of the loop of Henle, which results in low potassium levels (hypokalemia),[2] increased blood pH (alkalosis), and normal to low blood pressure. There are two types of Bartter syndrome: neonatal and classic. A closely associated disorder, Gitelman syndrome, is milder than both subtypes of Bartter syndrome.
| Bartter syndrome | |
|---|---|
| Other names | Salt-wasting nephropathy[1] |
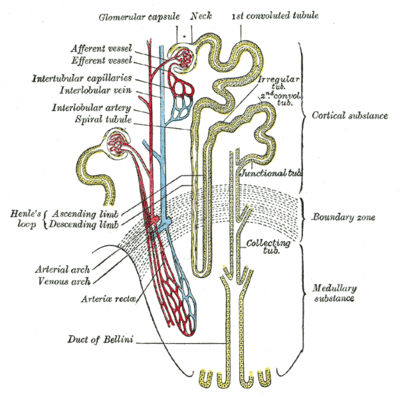 | |
| Scheme of renal tubule and its vascular supply. | |
| Specialty | Endocrinology |
Signs and symptoms
In 90% of cases, neonatal Bartter syndrome is seen between 24 and 30 weeks of gestation with excess amniotic fluid (polyhydramnios). After birth, the infant is seen to urinate and drink excessively (polyuria, and polydipsia, respectively). Life-threatening dehydration may result if the infant does not receive adequate fluids. About 85% of infants dispose of excess amounts of calcium in the urine (hypercalciuria) and kidneys (nephrocalcinosis), which may lead to kidney stones. In rare occasions, the infant may progress to kidney failure.
Patients with classic Bartter syndrome may have symptoms in the first two years of life, but they are usually diagnosed at school age or later. Like infants with the neonatal subtype, patients with classic Bartter syndrome also have polyuria, polydipsia, and a tendency to dehydration, but normal or just slightly increased urinary calcium excretion without the tendency to develop kidney stones. These patients also have vomiting and growth retardation. Kidney function is also normal if the disease is treated,[3] but occasionally patients proceed to end-stage kidney failure. Bartter syndrome consists of low levels of potassium in the blood, alkalosis, normal to low blood pressures, and elevated plasma renin and aldosterone. Numerous causes of this syndrome probably exist. Diagnostic pointers include high urinary potassium and chloride despite low serum values, increased plasma renin, hyperplasia of the juxtaglomerular apparatus on kidney biopsy, and careful exclusion of diuretic abuse. Excess production of prostaglandins by the kidneys is often found. Magnesium wasting may also occur. Homozygous patients suffer from severe hypercalciuria and nephrocalcinosis.[4]
Pathophysiology
Bartter syndrome is caused by mutations of genes encoding proteins that transport ions across renal cells in the thick ascending limb of the nephron also called as the ascending loop of Henle.[3] Specifically, mutations directly or indirectly involving the Na-K-2Cl cotransporter are key. The Na-K-2Cl cotransporter is involved in electroneutral transport of one sodium, one potassium, and two chloride ions across the apical membrane of the tubule. The basolateral calcium-sensing receptor has the ability to downregulate the activity of this transporter upon activation. Once transported into the tubule cells, sodium ions are actively transported across the basolateral membrane by Na+/K+-ATPases, and chloride ions pass by facilitated diffusion through basolateral chloride channels. Potassium, however, is able to diffuse back into the tubule lumen through apical potassium channels, returning a net positive charge to the lumen and establishing a positive voltage between the lumen and interstitial space. This charge gradient is obligatory for the paracellular reabsorption of both calcium and magnesium ions.
Proper function of all of these transporters is necessary for normal ion reabsorption along the thick ascending limb, and loss of any component can result in functional inactivation of the system as a whole and lead to the presentation of Bartter syndrome. Loss of function of this reabsorption system results in decreased sodium, potassium, and chloride reabsorption in the thick ascending limb, as well as abolishment of the lumen-positive voltage, resulting in decreased calcium and magnesium reabsorption. Loss of reabsorption of sodium here also has the undesired effect of abolishing the hypertonicity of the renal medulla, severely impairing the ability to reabsorb water later in the distal nephron and collecting duct system, leading to significant diuresis and the potential for volume depletion. Finally, increased sodium load to the distal nephron elicits compensatory reabsorption mechanisms, albeit at the expense of potassium by excretion by principal cells and resulting hypokalemia. This increased potassium excretion is partially compensated by α-intercalated cells at the expense of hydrogen ions, leading to metabolic alkalosis.
Bartter and Gitelman syndromes can be divided into different subtypes based on the genes involved:[5]
| Name | Bartter type | Associated gene mutations | Defect |
| neonatal Bartter's syndrome | type 1 | SLC12A1 (NKCC2) | Na-K-2Cl symporter |
| neonatal Bartter's syndrome | type 2 | ROMK/KCNJ1 | thick ascending limb K+ channel |
| classic Bartter's syndrome | type 3 | CLCNKB | Cl− channel |
| Bartter's syndrome with sensorineural deafness | type 4 | BSND[6] | Cl− channel accessory subunit |
| Bartter's syndrome associated with autosomal dominant hypocalcemia | type 5 | CASR[7] | activating mutation of the calcium-sensing receptor |
| Gitelman's syndrome | - | SLC12A3 (NCCT) | Sodium-chloride symporter |
Diagnosis
People suffering from Bartter syndrome present symptoms that are identical to those of patients who are on loop diuretics like furosemide, given that the loop diuretics target the exact transport protein that is defective in the syndrome (at least for type 1 Bartter syndrome). The other subtypes of the syndrome involve mutations in other transporters that result in functional loss of the target transporter. Patients often admit to a personal preference for salty foods.
The clinical findings characteristic of Bartter syndrome are hypokalemia, metabolic alkalosis, and normal to low blood pressure. These findings may also be caused by:
- Chronic vomiting: These patients will have low urine chloride levels; they have relatively higher urine chloride levels.
- Abuse of diuretic medications (water pills): The physician must screen urine for multiple diuretics before diagnosis is made.
- Magnesium deficiency and calcium deficiency: These patients will also have low serum and urine magnesium and calcium.
Patients with Bartter syndrome may also have elevated renin and aldosterone levels.[8]
Prenatal Bartter syndrome can be associated with polyhydramnios.[9]
Related conditions
- Bartter and Gitelman syndromes are both characterized by low levels of potassium and magnesium in the blood, normal to low blood pressure, and hypochloremic metabolic alkalosis.[10]
However, Bartter syndrome is also characterized by high renin, high aldosterone, hypercalciuria, and an abnormal Na+-K+-2Cl− transporter in the thick ascending limb of the loop of Henle, whereas Gitelman syndrome causes hypocalciuria and is due to an abnormal thiazide-sensitive transporter in the distal segment.
Pseudo-Bartter’s syndrome is a syndrome of similar presentation as Bartter syndrome but without any of its characteristic genetic defects. Pseudo-Bartter’s syndrome has been seen in cystic fibrosis,[11] as well as in excessive use of laxatives.[12]
Treatment
Medically supervised sodium, chloride and potassium supplementation is necessary, and spironolactone can be also used to reduce potassium loss.[2] Free and unqualified access to water is necessary to prevent dehydration, as patients maintain an appropriate thirst response.
In severe cases where supplementation alone cannot maintain biochemical homeostasis, nonsteroidal anti-inflammatory drugs (NSAIDs) can be used to reduce glomerular filtration, and can be very useful, although may cause gastric irritation and should be administered alongside stomach acid suppression therapies. Angiotensin-converting enzyme (ACE) inhibitors can also be used to reduce glomerular filtration rate. In young babies and children, a low threshold to check serum electrolytes during periods of illness compromising fluid intake in necessary.
Surveillance renal ultrasound should be employed to monitor for the development of nephrocalcinosis, a common complication which further augments urinary concentrating difficulty.
Prognosis
The limited prognostic information available suggests that early diagnosis and appropriate treatment of infants and young children with classic Bartter Syndrome may improve growth and perhaps intellectual development. On the other hand, sustained hypokalemia and hyperreninemia can cause progressive tubulointerstitial nephritis, resulting in end-stage kidney disease (kidney failure). With early treatment of the electrolyte imbalances, the prognosis for patients with classic Bartter Syndrome is good.
History
The condition is named after Dr. Frederic Bartter, who, along with Dr. Pacita Pronove, first described it in 1960 and in more patients in 1962.[8][13][14][15]
References
- "Bartter syndrome: MedlinePlus Medical Encyclopedia". medlineplus.gov. Retrieved 29 September 2019.
- "Bartter Syndrome: Tubular and Cystic Kidney Disorders: Merck Manual Home Edition". Archived from the original on 4 January 2008. Retrieved 2007-12-31.
- Rodriguez-Soriano J (1998). "Bartter and related syndromes: the puzzle is almost solved". Pediatr Nephrol. 12 (4): 315–27. doi:10.1007/s004670050461. PMID 9655365.
- http://ajprenal.physiology.org/content/307/9/F991.long
- Naesens M, Steels P, Verberckmoes R, Vanrenterghem Y, Kuypers D (2004). "Bartter's and Gitelman's syndromes: from gene to clinic". Nephron Physiol. 96 (3): 65–78. doi:10.1159/000076752. PMID 15056980.
- Zaffanello M, Taranta A, Palma A, Bettinelli A, Marseglia GL, Emma F (2006). "Type IV Bartter syndrome: report of two new cases". Pediatr. Nephrol. 21 (6): 766–70. doi:10.1007/s00467-006-0090-x. PMID 16583241.
- Vezzoli G, Arcidiacono T, Paloschi V, et al. (2006). "Autosomal dominant hypocalcemia with mild type 5 Bartter syndrome". J. Nephrol. 19 (4): 525–8. PMID 17048213.
- Bartter FC, Pronove P, Gill JR, MacCardle RC (1962). "Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome". Am J Med. 33 (6): 811–28. doi:10.1016/0002-9343(62)90214-0. PMID 13969763. Reproduced in Bartter FC, Pronove P, Gill JR, MacCardle RC (1998). "Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. 1962". J. Am. Soc. Nephrol. 9 (3): 516–28. PMID 9513916.
- Dane B, Yayla M, Dane C, Cetin A (2007). "Prenatal diagnosis of Bartter syndrome with biochemical examination of amniotic fluid: case report". Fetal Diagn. Ther. 22 (3): 206–8. doi:10.1159/000098719. PMID 17228161.
- Gitelman HJ, Graham JB, Welt LG (1966). "A new familial disorder characterized by hypokalemia and hypomagnesemia". Trans Assoc Am Physicians. 79: 221–35. PMID 5929460.
- Royal Brompton & Harefield Hospital > Pseudo-Bartter’s syndrome Retrieved Mars, 2011
- Metyas, Samy; Rouman, Heba; Arkfeld, Daniel G. (2010). "Pregnancy in a Patient With Gouty Arthritis Secondary to Pseudo-Bartter Syndrome". Journal of Clinical Rheumatology. 16 (5): 219–220. doi:10.1097/RHU.0b013e3181e9312a.
- Proesmans W (2006). "Threading through the mizmaze of Bartter syndrome". Pediatr. Nephrol. 21 (7): 896–902. doi:10.1007/s00467-006-0113-7. PMID 16773399.
- synd/2328 at Who Named It?
- "Bartter's syndrome". www.whonamedit.com.
External links
| Classification | |
|---|---|
| External resources |