Ring 18
Ring 18 is a genetic condition caused by a deletion of the two tips of chromosome 18 followed by the formation of a ring-shaped chromosome. It was first reported in 1964.[2]
| Ring 18 | |
|---|---|
| Other names | Ring chromosome 18[1] |
| Specialty | Medical genetics |
Signs and symptoms
Ring 18 causes a wide range of medical and developmental concerns.[3] As discussed above, people with ring 18 can have features of both distal 18q- and 18p-. The features of distal 18q- and 18p- vary greatly because of the variability of the deletion size and breakpoint locations between people.[4] Because ring 18 can involve unique deletions of both the p and q arms of the chromosome there is twice as much reason for the variability between individuals. This variation is also partly attributable to the incidence of mosaicism, which is relatively common in people with ring 18.
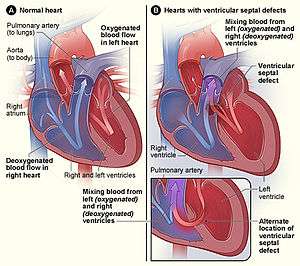
- Holoprosencephaly has been reported in some people with ring 18.[5] This is due to the deletion of the TGIF gene on the short arm of chromosome 18 in some people with ring 18.[6]Approximately 30-40% of people with ring 18 have a congenital heart anomaly. Septal defects are the most common type of defect reported in this population.
- Hypotonia is frequently seen in the ring 18 population. Seizures, though uncommon, have been reported in people with ring 18.
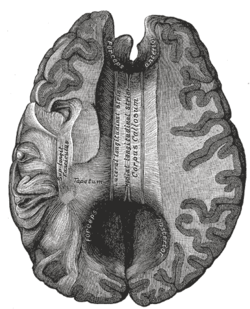
- In some children without “classic” holoprosencephaly, microforms of holoprosencephaly may be noted on MRI, including missing olfactory tracts and bulbs and absent or hypoplastic corpus callosum.
- Strabismus as well as nystagmus have both been reported in infants and children with ring 18.
- Hearing loss has been reported and may be related to ear canal atresia or stenosis.
- Umbilical and inguinal hernias have been reported in a small number of people with ring 18.
- Unilateral renal hypoplasia and aplasia have both been reported in individuals with ring 18. Hydronephrosis as well as pyelonephritis have also been reported in a few individuals. Cryptorchidism, hypospadias, and micropenis have been seen in males with ring 18, while females have been reported with hypoplastic labia.
- Foot abnormalities are common within the ring 18 population. Scoliosis as well as pectus excavatum have also been frequently reported.
- Several people with ring 18 have growth hormone deficiency. Hypothyroidism has also been reported in a minority of people.
- Cognitive ability varies; according to a literature review, the degree of impairment may fall anywhere between the mild and severe ends of the spectrum.
- Facial features of ring 18 include low-set, dysplastic ears, epicanthic folds, and hypertelorism. Micrognathia has also been reported.
Genetic
Individuals with ring 18 have one of their two copies of chromosome 18 that has formed the shape of a ring. The ring is formed when the caps on both the long arm (q) and the short arm (p) of one copy of chromosome 18 are lost and the new ends re-join to form the ring. Because the ring involves deletions of both the long arm (18q-) and the short arm (18p-) of chromosome 18, individuals with ring 18 can have features of both 18p- as well as distal 18q-.
Diagnosis
Suspicion of a chromosome abnormality is typically raised due to the presence of developmental delays or birth defects. Diagnosis of ring 18 is usually made via a blood sample. A routine chromosome analysis, or karyotype, is usually used to make the initial diagnosis, although it may also be made by microarray analysis. Increasingly, microarray analysis is also being used to clarify breakpoints. Prenatal diagnosis is possible via amniocentesis or chorionic villus sampling.
Treatment
At present, treatment for ring 18 is symptomatic, meaning that the focus is on treating the signs and symptoms of the conditions as they arise. To ensure early diagnosis and treatment, it is suggested that people with ring 18 undergo routine screenings for thyroid, hearing, and vision problems.
Research
Currently, research is focusing on identifying the role of the genes on 18p and 18q in causing the signs and symptoms associated with deletions of 18p and/or 18q. This will ultimately enable predictive genotyping. Thus far, several genes on chromosome 18 have been linked with a phenotypic effect.
TGIF - Mutations and deletions of this gene, which is located on18p, have been associated with holoprosencephaly. Penetrance is incomplete, meaning that a deletion of one copy of this gene is not in and of itself sufficient to cause holoprosencephaly. Ten to fifteen percent of people with 18p- have holoprosencephaly, suggesting that other genetic and environmental facts play a role in the etiology of holoprosencephaly in these individuals.
TCF4 – In 2007, deletions of or point mutations in this gene, which is located on 18q, were identified as the cause of Pitt-Hopkins disease. This is the first gene that has been definitively shown to directly cause a clinical phenotype when deleted. If a deletion includes the TCF4 gene (located at 52,889,562-52,946,887), features of Pitt-Hopkins may be present, including abnormal corpus callosum; short neck; small penis; accessory and wide-spaced nipples; broad or clubbed fingers; and sacral dimple. Those with deletions inclusive of TCF4 have a significantly more severe cognitive phenotype.
TSHZ1 - Point mutations and deletions of this gene, located on 18q, are linked with congenital aural atresia [7] Individuals with deletions inclusive of this gene have a 78% chance of having aural atresia.
Critical regions – Recent research has narrowed the critical regions for four features of the distal 18q- phenotype down to a small segment of distal 18q, although the precise genes responsible for those features remain to be identified.
| Feature | Critical Region | Chromosome Bands | Penetrance |
|---|---|---|---|
| Kidney malformation | 70,079,559-73,287,604 | 18q22.3-q23 | 25% |
| Dysmyelination | 71,669,548-73,287,604 | 18q22.3-q23 | 100% |
| Growth hormone response failure | 71,669,548-73,287,604 | 18q22.3-q23 | 90% |
Haplolethal Regions - There are two regions on chromosome 18 that has never been found to be deleted. They are located between the centromere and 22,826,284 bp (18q11.2) and between 43,832,732 and 45,297,446 bp (18q21.1). It is hypothesized that there are genes in these regions that are lethal when deleted.
Nomenclature
The phrase “ring 18” refers to the shape that the normally linear chromosome assumes when one tip of the chromosome joins the other. A ring-shaped chromosome is the result. In the case of ring 18, one of the two copies of chromosome 18 has formed a ring.
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Ring chromosome 18 syndrome". www.orpha.net. Retrieved 16 August 2019.
- Gropp et al (1964). Multiple congenital anomalies associated with a partially ring-shaped chromosome probably derived from chromosome no. 18 in man. Nature 202:829-30.
- http://www.chromosome18.org/TheConditions/Ring18/tabid/128/Default.aspx%5B%5D
- Heard PL, Carter EM, Crandall AC, Sebold C, Hale DE, Cody JD (July 2009). "High resolution genomic analysis of 18q- using oligo-microarray comparative genomic hybridization (aCGH)". Am. J. Med. Genet. A 149A (7): 1431–7.
- Yanoff et al (1970). Ocular and cerebral abnormalities in chromosome 18 deletion defect. Am J Ophthalmol 70(3):391-340.
- Solomon et al (2010). Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet 154C:133–41.
- Feenstra et al (2011). Disruption of teashirt zinc finger homeobox 1 is associated with congenital aural atresia in humans. Am J Hum Genet 89(6):813-9.