Ring chromosome 15
Ring chromosome 15 (denoted as r15 in humans) is a condition that arises when chromosome 15 fuses to form a ring. Usually, r15 forms due to modifications or deletion of genes on chromosome 15 in the preliminary stages of embryonic development but it can rarely also be inherited.[1]
| Ring chromosome 15 | |
|---|---|
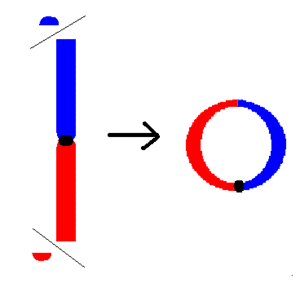 | |
| Formation of a ring | |
| Specialty | Medical genetics |
Ideally if there is no deletion at the ends of the chromosome and no loss of genetic material, the individual retains the normal appearance (phenotype) with relatively slight difference. However, when there is deletion of genetic information at the distal unstable ends and then the subtelomeric structures fuse, syndromes associated with that particular chromosome arise.[2] All chromosomes have the capacity to form such rings; the type and intensity of the symptoms depend on the amount of the genetic information lost. Thus, the treatment for r15 predominantly targets the eradication of these symptoms rather than the chromosome ring itself.
Presentation
As the cases reported are less in number, and the phenotype expression for ring chromosome 15 syndrome occurs over a wider spectrum; genes inside patients could have different levels of mosaicism.[3][4][5]
Common features[6], however, include growth delay, mental retardation and congenital malformation.
Of 25 studies examined by Butler MG,[3] all cases reported growth deficiency, 95% of cases showed varied levels of mental retardation, and 88% of the patients showed microcephaly.
Besides those top 3 symptoms that are found most frequently, other symptoms miscellaneous include: Delayed bone age (7%), Hypertelorism(46%), Brachydactyly(44%), Triangular face(42%), Speech delay(39%),Frontal bossing(36%), Anomalous ear(30%), Café-au-lait spots(30%), Cryptorchidism(30%) and Cardiac abnormalities(30%)[3]
A precise, concrete genotype-phenotype association hasn't been established yet but scientists currently have progressive understanding of the overarching rationale responsible for the phenotypes. For example, by applying FISH analysis, it was revealed that growth retardation (the most common phenotype) might be caused by terminal deletion of 15q26 (a specific region on the chromosome 15 long-arm).[7] Besides the mentioned symptoms found frequently in chromosome 15 ring patients, other co-occurring diseases exist that are predicted to be related to modification of chromosome 15.[2] These include: congenital diaphragmatic hernia, Russell-Silver syndrome, Prader-Willi syndrome and Autism.
Reported case
Since this is a rare disease, there are only a handful of reported cases. [2]
A 4.5 year old girl who was born at 36 weeks of gestation was found with this syndrome. Both her parents were normal and her mother abstained from drugs, smoking and alcohol during pregnancy. Distinct features she portrayed included: symmetrical growth retardation, cafe au lait spots on the chest, abdomen and thigh, Scarce hair, 5th finger clinodactyly, Delayed bone age (revealed by bone radiographing image technique) and Height and weight below 3rd percentile
The following hormones were observed at a normal level: Basal luteinizing hormone, Testosterone, Human Chorionic Gonadotropin (HCG) and Follicle Stimulating Hormone
Oestrogen levels were high
Mechanism
The human body stores its genetic material in chromosomes. The number of chromosomes and the gene locus on the chromosome is unique to each species. Humans have 23 pairs of chromosomes, 22 pairs of autosomal chromosomes and 1 pair of sex chromosomes that differentiate between males and females.[8]All human chromosomes have 2 arms - the p(short) and the q(long) arm which are separated by the centromeres.[9]
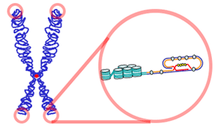
There are two proposed mechanisms of forming the ring chromosome. One suggests that the chromosome 15 undergoes distortion on both p and q arms prior to the fusion of two broken arms, resulting great loss of genetic materials. Therefore, the patients of this type of r15 display severe clinical features.[10]
Another method pivots upon the direct fusion of two telomeres without losing any of the telomeric and subtelomeric sequences. Consequently, most of the genetic materials is conserved and symptoms are expressed in a milder form, introducing convolution during diagnoses.[11]
Diagnosis
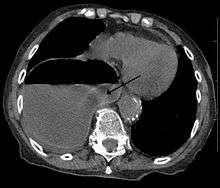
In most cases, postnatal diagnosis is done and up to 2011, only four cases are reported via prenatal diagnosis.[2] Congenital diaphragmatic hernia and intrauterine growth retardation(these two signs put the patients at the risk of afflicting with r15) by fetal ultrasound (Obstetric ultrasonography) at the time period of 16-24 weeks, further investigation and diagnostics (such as karyotyping) must be performed to test the possibility of ring chromosome 15.
Postnatal diagnosis
Patients could be considered to be at risk of developing ring chromosome 15 if they are found with:growth deficiency,[2] café-au-lait spots, bone age delay and simian crease or other dysmorphic features after birth.
For chromosome examination, some research suggests to conduct 72 hour peripheral lymphocytes’ culture, routinely stained with Quinacrine.[12](This is a way to prepare the chromosome and examine the karyotype)
Management
Not many studies have been published up to date regarding the accurate and efficient treatment of ring chromosome 15.[13]
Not many studies have been published up to date regarding the accurate and efficient treatment of ring chromosome 15, but a research article produced by scientists from The Children’s Hospital of Zhejiang University school of Medicine indicates that growth hormone(GH) might be one of the drugs to alleviate the symptoms of growth retardation. To treat this and emphasize the association, the scientists applied rhGH with a dose of 0.1 U/kg-1d-1 for four months, and a 3 cm increment of height with no remarkable side effects are observed.[2]
Epidemiology
The repeated emphasis on the rarity of the disease indicates that the incidence and distribution is fairly low; less than 50 cases were reported so far. Currently, there are only 3 cases reported in china.
Any of the 23 pairs of chromosomes can be ringed, and a recent study conducted by the 'Human Ring Chromosome Registry' in China revealed that the rather frequent forms of ring chromosomes reported were 13, 15, 18 and 22.[14]
In 2012, the first report of Ring Chromosome 22 was reported in India - a 5 year old male child with the karyotype 46, XY, r(22)[15]
From these variety of cases reported, it can be concluded that ring chromosome syndromes are rare congenital disorders that are likely to occur in both males and females, and the symptoms can be observed from birth since most of the time it arises during the embryonic stage. All races and ethnicities are prone to the disorders and the risk can be higher if the parents are carriers since it is genetically inherited.
The epidemiologic data may not be completely accurate as it is still unclear whether or not all the clinical diagnostic manifestations are pathological to ring chromosomes. Since the symptoms are far too many and highly non-specific, there are very high chances of misdiagnoses.
History
r15 is an uncommon genetic disorder first spotted by Dr Petrea Jacobsen in 1966.[16]Despite the rarity of this disease, it was discovered more than half a century ago. Until date, only around 40 cases have been reported.[13]
References
- "Ring chromosome 15 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2019-04-11.
- Xu, F; Zou, CC; Liang, L; Huang, XM; Shao, YN (2011). "Ring Chromosome 15 Syndrome:Case Report and Literature Review". HK J Paediatr (new series): 175–179.
- Butler, Merlin G.; Fogo, Agnes B.; Fuchs, David A.; Collins, Francis S.; Dev, Viathilingam G.; Phillips, John A.; Optiz, John M.; Reynolds, James F. (Jan 1988). "Two patients with ring chromosome 15 syndrome". American Journal of Medical Genetics. 29 (1): 149–154. doi:10.1002/ajmg.1320290119. ISSN 0148-7299. PMC 5083070.
- Roback, Ellen W.; Barakat, Amin J.; Dev, V. G.; Mbikay, Majambu; Chrétien, Michel; Butler, Merlin G. (1991-01-01). "An infant with deletion of the distal long arm of chromosome 15 (q26.1→qter) and loss of insulin-like growth factor 1 receptor gene". American Journal of Medical Genetics. 38 (1): 74–79. doi:10.1002/ajmg.1320380117. ISSN 0148-7299. PMC 5493390.
- Nuutinen, M; Kouvalainen, K; Knip, M (1995-06-01). "Good growth response to growth hormone treatment in the ring chromosome 15 syndrome". Journal of Medical Genetics. 32 (6): 486–487. doi:10.1136/jmg.32.6.486. ISSN 1468-6244.
- Peoples, R.; Milatovich, A.; Francke, U. (1995). "Hemizygosity at the insulin-like growth factor I eceptor (IGF1R) locus and growth failure in the ring chromosome 15 syndrome". Cytogenetic and Genome Research. 70 (3–4): 228–234. doi:10.1159/000134040. ISSN 1424-859X.
- Harada, N; Shimokawa, O; Nagai, T; Kato, R; Kondoh, T; Niikawa, N; Matsumoto, N (2002-10-08). "A 4-Mb critical region for intrauterine growth retardation at 15q26". Clinical Genetics. 62 (4): 340–342. doi:10.1034/j.1399-0004.2002.620416.x. ISSN 0009-9163.
- Reference, Genetics Home. "What is a chromosome?". Genetics Home Reference. Retrieved 2019-04-11.
- "Definition of Long arm of a chromosome". MedicineNet. Retrieved 2019-04-11.
- Kosztolanyi, G (Feb 1987). "Does "ring syndrome" exist? An analysis of 207 case reports on patients with a ring autosome". Human Genetics. 75 (2): 174–179. doi:10.1007/bf00591082. ISSN 0340-6717.
- Pezzolo, Annalisa; Gimelli, Giorgio; Cohen, Amnon; Lavaggetto, Antonella; Romano, Cesare; Fogu, Giuseppina; Zuffardi, Orsetta (Aug 1993). "Presence of telomeric and subtelomeric sequences at the fusion points of ring chromosomes indicates that the ring syndrome is caused by ring instability". Human Genetics. 92 (1): 23–27. doi:10.1007/bf00216140. ISSN 0340-6717.
- Hitayezu, J. Uwineza, A. Murorunkweri, S. Ndinkabandi, J. Mutesa, L. (2013-09-14). A Case of Rwandan Patient with Ring Chromosome 15 Syndrome. Rwanda Health Communication Center - Rwanda Biomedical Center (RHCC - RBC). OCLC 863408307.CS1 maint: multiple names: authors list (link)
- Glass, Ian A.; Rauen, Katherine A.; Chen, Emily; Parkes, Jillian; Alberston, Donna G.; Pinkel, Daniel; Cotter, Philip D. (2005-11-03). "Ring chromosome 15: characterization by array CGH". Human Genetics. 118 (5): 611–617. doi:10.1007/s00439-005-0030-z. ISSN 0340-6717.
- Hu, Qiping; Chai, Hongyan; Shu, Wei; Li, Peining (2018-02-27). "Human ring chromosome registry for cases in the Chinese population: re-emphasizing Cytogenomic and clinical heterogeneity and reviewing diagnostic and treatment strategies". Molecular Cytogenetics. 11 (1). doi:10.1186/s13039-018-0367-3. ISSN 1755-8166.
- Mahajan, S; Kaur, A; Singh, J (2012-01-01). "Ring Chromosome 22: A Review of the Literature and First Report from India". Balkan Journal of Medical Genetics. 15 (1): 55–59. doi:10.2478/v10034-012-0009-8. ISSN 1311-0160.
- JACOBSEN, PETREA (2009-09-02). "A RING CHROMOSOME IN THE 13-15 GROUP ASSOCIATED WITH MICROCEPHALIC DWARFISM, MENTAL RETARDATION AND EMOTIONAL IMMATURITY". Hereditas. 55 (2–3): 188–191. doi:10.1111/j.1601-5223.1966.tb02047.x. ISSN 0018-0661.