

Chapter 10: PCR for Detection and Characterization of Bacterial Meningitis Pathogens: Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae
 ShareCompartir
ShareCompartir
Printer friendly version [56 pages]
- Overview of PCR technologies
In developing countries, the most commonly used approaches for detection and characterization of bacterial meningitis pathogens include culture, Gram stain, and latex agglutination. Although culture is considered the gold standard for case confirmation in clinics, the positive rate is relatively low due to suboptimal storage and transportation conditions, culture practice, and/or antibiotic treatment administered before the specimen is collected. While Gram staining is important, inexpensive, and should be performed whenever possible, it merely gives a clue as to the genus and species of the etiological agent. The reading of latex agglutination results is subjective and can be difficult to interpret, especially when a specimen's bacterial load is low. It is also not feasible to do quality control on latex agglutination. Culture should be kept as the gold standard as cultured bacteria are sources of data for antibiotic susceptibility, complete subtyping, the expression of antigens that are to be included in future vaccines, and pathophysiology of isolates. Specimens that do not yield any culture can still be analyzed by molecular methods (see below) that can be applied on DNA extracted from clinical specimens (typically, blood and CSF).
- Polymerase chain reaction (PCR)
PCR was developed in the mid- to late 1980s (36, 42) and is considered one of the most important methodological inventions in molecular biology. It is designed to permit selective amplification of a specific target DNA sequence(s) within a heterogeneous collection of DNA sequences (e.g., total genomic DNA). In PCR, the DNA target is exponentially amplified through repeating three major steps: 1) denaturation of double-stranded DNA into single-stranded DNA; 2) annealing of primers to the complementary single-stranded target sequences; and 3) extension of the primers in the 5' to 3' direction by heat-stable DNA polymerase to produce double-stranded DNA molecules. The copy number of DNA molecules is doubled in each extension step, generating millions of copies of the original DNA molecules when PCR is completed. Because the method does not require live or intact cells, PCR is a valuable tool for detecting bacterial pathogenic agents from clinical specimens, where bacteria die or lyse easily due to inappropriate storage conditions or prior antibiotic treatment. PCR is now widely used in the diagnosis and surveillance of bacterial pathogens because of its high sensitivity and specificity and high throughput capabilities. It provides a complementary tool to classic phenotype-based methods such as culture, Gram stain, and latex agglutination and often enhances confirmatory results (3).
A number of conventional PCR assays have been developed for detection and subtyping of bacterial and viral pathogens. Conventional PCR detects products at the end point of DNA amplification by visualizing amplicons using agarose gel electrophoresis. Gel-based detection requires that tubes containing PCR amplicons be opened and manipulated, thereby greatly increasing the risk of contamination of laboratory space, equipment, and reagents with amplified materials. Conventional PCR is also very time consuming and less sensitive and specific than a type of PCR called real-time PCR, so it is primarily used for typing assays employing purified cultures or clinical specimens that contain the organism in high density. The use of real-time PCR is rapidly expanding because it is easier to perform and, being a closed system, it reduces potential contamination problems inherent to conventional PCR. Many of the same precautions mentioned for conventional PCR assays also apply to real-time PCR assays.
- Real-time PCR technology
Real-time PCR is also known as quantitative real-time polymerase chain reaction (Q-PCR/qPCR) or kinetic polymerase chain reaction, which combines amplification and detection in one step through the use of fluorescent dyes. There are two types of detection systems: non-specific and specific. Non-specific detection systems use a fluorescent dye that intercalates into any double-stranded DNA molecules and emits enhanced fluorescence. This detection system is relatively inexpensive but susceptible to false positivity. Specific detection systems rely on fluorescent resonance energy transfer probes that specifically recognize target sequences, thus making them the systems of choice for the molecular detection assays described here. Specific detection systems are more expensive than non-specific detection systems and require sophisticated probe designs. Three types of probes are currently in use, including: hydrolysis, hybridization, and hairpin probes (7, 13). A fluorescent signal is only generated if the probe interacts with its specific target and is subsequently hydrolyzed during amplification. The resulting increase in fluorescence is proportional to the amount of amplified PCR product in the reaction.
The first use of dual-labeled hydrolysis probes was reported in 1993 (30) and has been widely used in many laboratories since that time. A dual-labeled hydrolysis probe is an oligonucleotide (~17-35 bp long) labeled with a reporter fluorophore (usually a short wavelength colored dye) at the 5' end and a quencher fluorophore (usually a long wavelength colored dye) at the 3' end or at an internal thymine or "T" residue. Optimally, the quencher dye should be 7-15 base pairs from the reporter dye. When the probe is intact and excited by a light source, the fluorescence emission of the reporter (or donor) dye is absorbed by the quencher (or acceptor) dye as a result of the close proximity of the dyes. This process is also known as fluorescence resonance energy transfer (FRET). During PCR amplification, the probe anneals to an internal region of the target DNA template between the forward and reverse primer. When DNA polymerase catalyzes the extension of the primer and reaches the region where the probe is bound, the 5' exonuclease activity of the DNA polymerase cleaves the probe and releases the reporter from the quencher. This allows emission of the fluorescence from the reporter dye to be observed because it is no longer absorbed by the quencher which has diffused away (Figure 1). The increase in fluorescence is proportional to the amount of amplified PCR product in the reaction and is measured cumulatively over the course of the entire PCR run (7, 30).
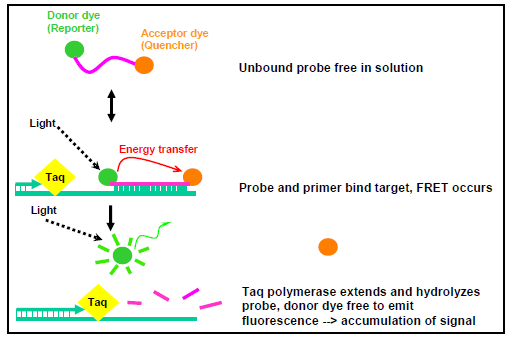
Figure 1. Chemistry of a dual-labeled hydrolysis probe...view larger
- Polymerase chain reaction (PCR)
- Target gene assays for detection and characterization of bacterial meningitis etiologies
Over the past several years, conventional and real-time PCR assays have been developed for detection of bacterial meningitis pathogens. Reliable assays have been extensively evaluated using invasive clinical isolates and/or clinical specimens from around the world (8, 12, 15, 17, 29, 35, 52, 60). In general, validated assays should have high sensitivity and specificity. They can be used as complementary approaches in bacterial disease diagnosis. The gene targets, DNA sequence of primers and probes, and the final concentration used in the PCR reactions described here to detect and characterize N. meningitidis, H. influenzae, and S. pneumoniae are listed in Tables 2-5.
The PCR strategy typically employed to detect the causative agent in a suspected case of bacterial meningitis is to first run each of the species-specific assays concurrently on the DNA extracted from the clinical specimen or isolate. The appropriate serogroup/serotype specific assays should then be run on any positive specimens. See Section V below, "Workflow for detection of bacterial meningitis pathogens by PCR", for more details.
- Species-specific real-time PCR assays
PCR detection of N. meningitidis, H. influenzae, and S. pneumoniae can be achieved by amplification of several potential gene targets (8, 35, 53, 60). The following assays have been developed and validated to be used on DNA extracted from clinical specimens (typically, blood and CSF) and bacterial isolates.
N. meningitidis
Two genes can be targeted in N. meningitidis species-specific assays, ctrA and sodC. The capsule transport to cell surface gene, ctrA, is highly conserved among isolates responsible for invasive meningococcal infections and has been used in both real-time and conventional PCR to detect N. meningitidis (35). It is a gene within the capsule locus (Figure 2). However, since at least 16% of carried meningococci lack ctrA (10, 14, 41), a real-time PCR assay to detect all meningococci, regardless of encapsulation status, was recently developed and validated (15). This assay targets the Cu, Zn superoxide dismutase gene, sodC, which is not genetically linked to the capsule locus. The sodC assay detects encapsulated meningococci, but it is also useful for detecting nongroupable meningococci that do not contain an intact ctrA, as will be recovered during carriage studies. For this reason, it is recommended that sodC be used for detection of N. meningitidis, if possible. sodC and ctrA primers and probes are listed in Table 2.
H. influenzae
The protein D encoding gene, hpd, encodes protein D, a highly conserved, surface-exposed lipoprotein that is present in all encapsulated and non-encapsulated H. influenzae (24, 45). The conserved nature of this gene and its presence in all strains of H. influenzae characterized to date make it a highly attractive gene target for the development of a H. influenzae species-specific real-time PCR assay. The recently developed and validated hpd real-time PCR assay is capable of detecting all six serotypes (a-f) and nontypeable (HiNT) H. influenzae with high sensitivity and specificity (60). Real-time PCR assays targeting bexA were developed and distributed because bexA is present in all six serotypes of H. influenzae. However, though sensitive for detection of Hib, it is less sensitive for Hia, Hic, and Hid, and does not detect Hie, Hif, or HiNT and should no longer be used. The primers and probes for the hpd assay are listed in Table 2.
S. pneumoniae
Both conventional and real-time PCR assays have been developed for the detection of S. pneumoniae, and target genes have included the pneumolysin (ply), autolysin (lytA), and pneumococcal surface adhesion (psaA) genes (8). However, false-positive results with ply-based PCR have been reported when applied to upper respiratory tract specimens. A suggested explanation for these false positives is the detection of non-pneumococcal alpha-hemolytic streptococci (37, 47), which are normally present in the respiratory flora (e.g., Streptococcus mitis group and Streptococcus oralis) which sometimes contain a ply gene (62). The PCR detection assay for S. pneumoniae using a specific segment of the autolysin gene (lytA) is recommended because it is highly conserved within the species and it has been shown that this assay bestseparates S. pneumoniae from the genotypically similar species S. mitis, S. oralis, and S. pseudopneumoniae (33). The real-time PCR assay lytA primers and probes that have been found to be extremely reliable for detection of S. pneumoniae are listed in Table 2. Due to recombination events that occur between pneumococci and closely related streptococci, there will probably be rare false-positives or false-negatives for virtually any real-time assay for pneumococcal identification.
- Serogroup/serotype-specific real-time PCR assays
The capsule gene loci of both N. meningitidis and H. influenzae have areas that are both unique and conserved within each serogroup (N. meningitidis) or serotype (H. influenzae) thus providing gene targets for the development or real-time PCR assays designed to identify each specific serogroup or serotype.
N. meningitidis
N. meningitidis is classified into 12 serogroups on the basis of the chemical composition and linkage type of saccharide subunits of the capsular polysaccharide that are expressed on the bacterial cell surface. Major disease-causing serogroups include A, B, C, Y, and W135, the latter four of which produce sialic acid containing capsular polysaccharides; whereas serogroup A produces a poly-α1-6-linked N-acetylmannosamine 6-phosphate capsule (31). Outbreaks caused by serogroup X meningococci, which express poly-α1-4-linked N-acetylglucosamine 1-phosphate capsule (6) have also been reported (2, 20). Serogroup D is no longer recognized as a serogroup of N. meningitidis.
As illustrated in Figure 2, the genetic organization of the capsule locus is conserved among the serogroups. The capsule expression genes are located in four operons: one that encodes capsule biosynthesis (called syn or sia genes, depending on which nomenclature system is used) and three that encode the capsule transport to the cell surface proteins (ctr). The gene products of the ctr operon share high similarity with the ATP-dependent transporters of the ABC family (19) and are highly conserved among the major disease-causing serogroups (1, 18, 39, 55) and serogroup X (56). Sensitive real-time PCR assays targeting ctrA, which is the first gene in the capsule transport operon, have been developed for detection of all encapsulated and some non-encapsulated (nongroupable) N. meningitidis (12, 35), though a more specific and sensitive assay using the sodC gene as a target has been developed. The genetic differences among the capsule biosynthesis operons of meningococcal serogroups have facilitated the development of real-time PCR assays targeting serogroup-specific genes for capsule biosynthesis to determine the capsule genotype of a meningococcal isolate (35).
The gene sia (for sialic acid biosynthesis (16, 22), also called syn forcapsule biosynthesis (21, 51), are used for genotyping for serogroups B (synD), C (synE), Y (synF) and W135 (synG). The sacB gene is targeted for serogroup A and the xcbA gene, which most likely encodes the capsule polymerase, is targeted for serogroup X (2, 35). These target genes are depicted within the structure of the capsule gene complex for serogroups A, B, C, Y, W135, and X (Figure 2). The most current adapted primer and probe sequences for the serogrouping real-time PCR assays are listed in Table 3, though, periodically, the primers and probes are adapted as new information regarding probe chemistries and allelic variations become available.
There have been several different systems in place for naming the genes for meningococcal capsule biosynthesis. Some groups have called this operon syn for capsule biosynthesis (21, 51); other groups have used the sia nomenclature for sialic acid biosynthesis (16, 22); still others have referred to them as neu genes based on homologies to E. coli K1 genes for N-acetylneuraminic acidbiosynthesis (21). While the siaD genes of serogroups B, C, W135, and Y were initially thought to be alleles, more extensive sequencing analysis demonstrated that this is not so. The synD and synE genes of serogroups B and C, respectively, are alleles and encode capsular polysaccharide polymerases that catalyze different linkages of sialic acid monomers (α2→8 linkage for serogroup B and α2→9 linkage for serogroup C). However, the Y and W135 genes are over twice the size of the B and C genes and differ in nucleotide sequence (11, 50), though they are highly similar to each other. In addition, the polymerases for serogroups Y and W135 link heteropolymers of sialic acid plus either glucose or galactose, respectively. Thus, the capsular polysaccharide polymerase genes of serogroups Y and W135 are alleles. To continue to call all of these polymerase genes siaD would be a misnomer. For these reasons and for simplicity, this text will use the synABCD/E/F/G nomenclature listed above and in Table I.
Table 1. Serogroup capsule type and gene targets for genotyping real-time PCR assays
Sero-group Capsule type Gene Target Name Alternate Gene Names Ref A (α1→6)-N-acetyl-D-mannosamine-1-phosphate sacB (31) B (α2→8)- N-acetylneuraminic acid synD siaD
siaD of B
siaDB(5, 9, 23, 48, 61) C (α2→9)- N-acetylneuraminic acid synE siaD of C
siaDC(5, 9, 50, 58, 61) W135 6-D-Gal(α1→4)-N-acetylneuraminic acid(α2→6 synG siaD of W135
siaDW(4, 9, 14, 35, 49, 61) X (α1→4)-N-acetyl-D-glucosamine-1-phosphate xcbB (2, 6) Y 6-D-Glc(α1→4)-N-acetylneuraminic acid(α2→6) synF siaD of Y
siaDY(4, 9, 14, 35, 49, 61) For serogroups B, C, Y and W135, the first three genes of the syn operon encode functions for the synthesis of capsule polysaccharide precursors (32, 48, 51). The fourth gene product is a polymerase that catalyzes the formation of polymers with the serogroup-specific linkage. In serogroups B and C, the products of a four-gene operon (synABC plus the polysialyltransferase gene synD [Nmen B] or synE [Nmen C]) are responsible for biosynthesis of the sialic acid (also known as N-acetylneuraminic acid, NeuNAc, or NANA) homopolymer. Expression of the poly-N-acetylmamosamine-1-phosphate capsule polymer of serogroup A requires the sacABCD operon (formerly known as mynABCD (49), while expression of the poly-N-acetyl-D-glucosamine-1-phosphate capsule of serogroup X requires the xcbABC operon.
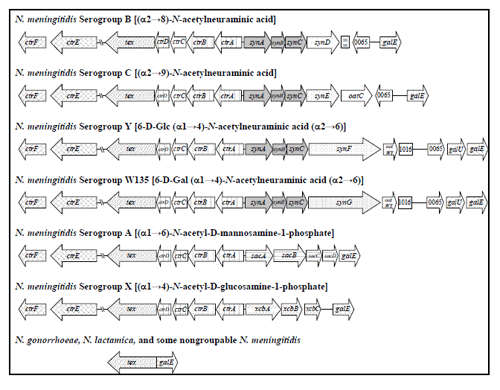
Figure 2. Genetic maps of the capsule gene complex (cps) of N. meningitidis (adapted from (14)). The ctrABCD operon encodes ATP-dependent export proteins (grid pattern). synABC (solid gray) D/E/F/G (dotted), sacABCD (horizontal striped), and xcbABC (dashed upward diagonal), encode the serogroup-specific enzymes for capsule polymer biosynthesis. oatC (serogroup C) and oatWY (serogroups W135 and Y), are co-transcribed with the syn operons and encode O-acetyltransferases (9). lipA and lipB code for proteins that were originally proposed to add a phospholipid-anchoring group onto the polysaccharide reducing end before transport (19). ctrE and ctrF (diagonal brick), formerly known as lipA and lipB, respectively, are involved in capsule transport (57). These gene products were once thought to be involved in post-polymerization modification. Many nongroupable, carried meningococci lack all or part of the capsule locus (10, 14); those lacking the entire locus have a genetic configuration at this position like those of N. gonorrhoeae and N. lactamica, which are not known to synthesize capsule....view larger
Haemophilus influenzae
The capsule locus of all six serotypes of H. influenzae (Hi a, b, c, d, e, and f) consists of three regions encoding functions for capsule polysaccharide synthesis, modification, and translocation (Figures 3 and 4) (25, 43, 44). bexDCBA in the ATP-driven export region (also known as Region I) code for protein components of an ATP-driven polysaccharide export apparatus. hcsA and hcsB in post polymerization modification region (also known as Region III) share high similarity with lipA and lipB (recently renamed ctrE and ctrF), respectively, which are involved in modification and export of meningococcal capsule polysaccharide (43). Both regions are common to all serotypes. The same nomenclature is used for genes in the two regions for all six serotypes. The serotype-specific region (previously Region II) contains genes for capsule synthesis and is unique to each serotype. The serotype-specific genes are named acs, bcs, etc. for "a capsule synthesis", "b capsule synthesis", and so on.
The cap locus encodes functions for H. influenzae capsule synthesis. The genetic organization of the cap locus has been well characterized in Hib and Hif, which belong to two phylogenetic divisions that are defined by multilocus enzyme electrophoresis typing. The ATP-driven export region includes most of Hia and Hib strains, and all of Hic, Hid, and Hie strains. Strains from this region have at least one completed cap locus flanked by insertion sequence (IS) element IS1016, except Hie. The majority of Hib strains and some Hia strains from this region have the direct-repeat configuration with the second copy of the bexA gene partially deleted as illustrated in Figure 4. The truncated cap locus is not required for capsule synthesis (26-28). In Hif, Hib, and Hia strains of the serotype-specific region and some Hie strains of the ATP-driven export region, the cap locus is flanked by sodC and HI1637 (26, 44).
While serotype b causes the vast majority of H. influenzae disease in countries without a Hib vaccination program, serotypes a, c, d, e, and f, and nontypeable H. influenzae (NTHi), also contribute to case numbers (43, 59). As implementation of Hib vaccine becomes more widespread, it is important to monitor incoming specimens for both b and non-b serotypes of H. influenzae. Therefore, serotype-specific real-time PCR assays have been developed that target the serotype-specific region genes where possible, or the 5' end of bexD, which is less conserved among the serotypes compared with the other genes in the export region. The genes targeted for real-time PCR assays specific to each serotype are as follows: acsB (Hia), bcsB (Hib), ccsD (Hic), dcsE (Hid), ecsH (Hie), and bexD (Hif) (Figure 3). The primer and probe information for each of these serotyping assays can be found in Table 4. Each of these assays has been shown to be highly specific and sensitive for their respective serotypes.
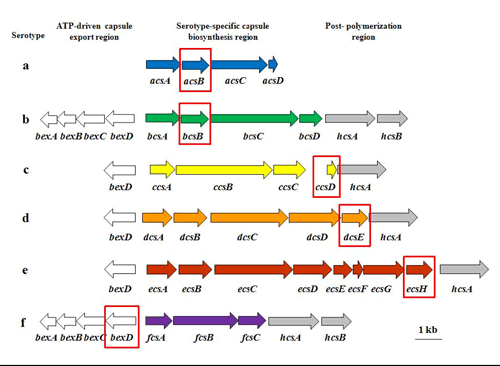
Figure 3. Capsule loci for H. influenzae serotypes a, b, c, d, e, and f, including the target genes for serotype-specific real-time PCR assays. The capsule locus of all six serotypes of H. influenzae (Hia-f) consists of three regions encoding functions for capsule polysaccharide synthesis, modification, and translocation. bexDCBA in the ATP-driven export region (white arrows) code for protein components of an ATP-driven polysaccharide export apparatus. hcsA and hcsB are in the post polymerization modification region (gray arrows) and may be involved in the modification and export of capsule polysaccharide. The serotype-specific region (colored arrows) contains genes for capsule synthesis and is unique to each serotype. The serotype-specific genes are named acs, bcs, etc. for "a capsule synthesis", "b capsule synthesis", and so on. With the exception of the Hif serotype-specific assay, the target genes for the serotype-specific assays can be found in this region and are highlighted by the red boxes...view larger
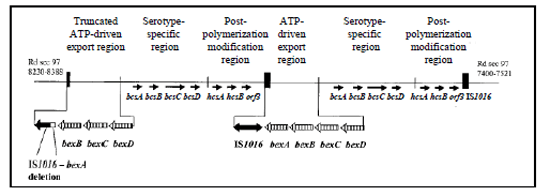
Figure 4. Genetic organization of the cap locus in Hib (adapted from (43)). Partially duplicated cap locus of Hib Hi 1007 showing the truncated ATP-driven export region with the 1.2-kb deletion between IS1016 and bexA...view larger
- S. pneumoniae serotyping PCR assays
S. pneumoniae can be further classified into at least 93 serotypes based on the immunochemistry of their capsular polysaccharides. The high cost of antisera, subjectivity in interpretation, need for a complete set of control strains, and technical expertise requirements associated with these serologic methods have resulted in the more recent development of PCR-based serotyping systems. PCR-serotyping has the potential to overcome some of the difficulties associated with serologic testing and the development of PCR-based assays for direct detection of serotypes from clinical specimens is a valuable aid in surveillance, particularly in situations where culture is insensitive. PCR assays (both conventional and real-time) for the detection of the more common serotypes are being developed and are discussed in more detail below (38, 54).
Multiplex conventional PCR assays for serotyping S. pneumoniae
A multiplex PCR-based serotyping scheme that includes 40 serotype specificities has been developed (38). This PCR approach has the potential to greatly reduce reliance upon conventional serotyping and provides serotype-determining potential to laboratories that lack type-specific antisera and other reagents needed for conventional serotyping, yet have the equipment necessary for DNA amplification and electrophoresis.
The multiplex approach uses 9 reactions to identify 40 serospecificities (Table 5 for primers, Tables 6-8 for schemes, and Figure 5 for PCR products) but also provides some flexibility that allows for altering combinations of serotypes included in each sequential reaction. These can be modified based on the most prevalent serotypes in any given geographic region but do require validation to ensure no cross reaction between serotype primer sets. Three such schemes based upon pneumococcal serotype prevalence in the USA, Africa, and Latin America have been designed (Tables 6-8). These schemes will continue to be refined as additional serotypes are added and primer sets updated to improve specificity and sensitivity. The most current methods are described at the CDC Streptococcus website and should be consulted on a regular basis
Real-time PCR assays for serotyping S. pneumoniae
A number of real-time PCR assays for serotyping S. pneumoniae have been published in the literature and others are being developed (34, 40). These real-time assays are recommended for determining serotypes from clinical specimens when DNA may be present in low amounts and insufficient for conventional multiplex PCR serotyping. Please see the Streptococcus Laboratory website for an example.
- Multiplex real-time PCR for pathogen detection
Real-time PCR allows for development of multiplex assays for detection of several genes in the same reaction by using specific probes with different fluorescent dye labels. Multiplex real-time PCR assays are available for detection of N. meningitidis, H. influenzae, and S. pneumoniae in a single reaction. In addition, assays for serogrouping N. meningitidis and serotyping S. pneumoniae using a multiplex approach have also been developed. However, they are not recommended for routine use. The multiplex approach requires careful optimization. Changes in DNA polymerase or PCR reagent concentrations may lead to loss of sensitivity. The assays need to be optimized and re-evaluated, which is a very time-consuming process and requires a complete set of reference strains.
- Species-specific real-time PCR assays
- Preparation of DNA template for PCR
- General considerations
Both crude DNA preps (boiled cell suspension) and extracted genomic DNA can be used as template for PCR amplification. DNA extraction from clinical specimens or isolates should be performed in a separate room from the room used for PCR reaction assembly (prior to DNA addition). If separate rooms are not possible, separate laboratory benches should be used. The use of boiled extracts may limit the risk of contamination as it limits manipulation. Moreover, PCR on clinical specimens should be performed in a separate room from where bacteria are cultured with strict organization of the laboratory workflow. Use of a biological safety cabinet is necessary for infectious materials such as clinical isolates, blood, and CSF. Separate pipettes, laboratory coats, and gloves should be used for performing DNA extraction procedures. Signage indicating which spaces and equipment are DNA-free and which are used to prepare or manipulate DNA would be helpful. UV irradiation and decontamination of surfaces and equipment with 10% bleach followed by 70% ethanol should be performed after any manipulation of nucleic acids at the laboratory bench. Always use filter-barrier pipette tips and change and discard gloves frequently.
- Equipment, consumables, and reagents for DNA extraction
- Equipment
- Microcentrifuge with refrigerating function
- Water bath or dry block heater
- Vortexer
- Freezer
- Refrigerator
- pH meter
- Balance
- Stir plate
- Consumables:
- 10% bleach (10:1, water: concentrated bleach) (make fresh weekly)
- 70% ethanol
- 1.5 ml microcentrifuge tubes (sterile, DNAase free, or PCR grade)
- 1 set of micropipettors (1-10 µl, 2-20 µl, 20-200 µl, and 100-1000 µl)
- Pre-sterilized filter tips (10 µl, 200 µl, and 1000 µl)
- Reagents:
- TE Buffer (10 mM Tris HCl, pH 8.0, 1 mM EDTA)
- Tris buffer (10 mM Tris HCl, pH 8.0)
- Lysozyme
- Mutanolysin
- Proteinase K (20 mg/ml)
- Lysis buffer (4% SDS, 10 mM EDTA pH 8.0)
- Digestion buffer
- Phenol
- Chloroform
- Phenol: chloroform (1:1)
- Commercial DNA extraction kits are available for culture and blood and body fluids
- Commercial PCR Master Mix containing dNTPs and DNA polymerase is available
- Equipment
- Preparing reagent solutions
EDTA, 0.1 M, pH 8.0 (100ml)
- Dissolve 3.7 g EDTA in 70 ml distilled deionized H2O (ddH2O).
- Adjust pH to 8.0 with 10 M NaOH.
- Add ddH2O to 100 ml.
- Autoclave at 121°C for 20 minutes.
- Store at room temperature.
Tris-HCl, 0.1 M, pH 8.0
- Dissolve 1.2 g Tris base in 80 ml ddH2O.
- Adjust to pH 8.0 with concentrated HCl.
- Mix and add ddH2O to 100 ml.
- Autoclave at 121°C for 20 minutes.
- Store at room temperature.
Tris-HCl, 0.1 M, pH 7.6
- Dissolve 1.2 g Tris base in 80 ml ddH2O.
- Adjust to pH 7.6 with concentrated HCl.
- Mix and add ddH2O to 100 ml.
- Autoclave at 121°C for 20 minutes.
- Store at room temperature.
20% Sodium dodecyl sulfate (SDS)*
- Dissolve 20 g SDS in 100 ml sterile ddH2O.
- Place the container into a 50-607°C water bath to facilitate dissolving.
- Avoid vigorous shaking that generates bubbles.
- Store at room temperature.
*Eye and respiratory protection should be worn when weighing powdered SDS.
TE buffer (10 mM Tris HCl, pH 8.0, 1 mM EDTA)
- Add 10 ml of 0.1 M Tris-HCl, pH 8.0.
- Add 1 ml of 0.1 M EDTA, pH 8.0.
- Add sterile ddH2O to 100 ml and mix well.
- Store at room temperature.
Tris-HCl buffer (10 mM, pH 8.0)
- Add 10 ml of 0.1 M Tris-HCl.
- Add 90 ml sterile ddH2O and mix well.
- Store at room temperature.
Lysis buffer (4% SDS, 10 mM EDTA pH 8.0)
- Add 20 ml of 20% SDS.
- Add 10 ml of 0.1 M EDTA pH 8.0.
- Add sterile ddH2O to 100 ml and mix well.
- Store at room temperature.
Mutanolysin stock solution (2,500 U/ml)
- Reconstitute the entire bottle of mutanolysin with sterile ddH2O to produce a concentration of 2,500 U/ml.
- Aliquot into sterile screw-top microcentrifuge tubes.
- Store aliquots at -20°C.
Digestion buffer (0.04 g/ml lysozyme and 75 U/ml mutanolysin in TE buffer)
- For 1 ml of TE buffer containing lysozyme and mutanolysin, add 40 mg lyophilized lysozyme to a 1.5 ml microcentrifuge tube.
- Add 1 ml TE buffer.
- Add 30 µl of a stock solution of mutanolysin at 2500 U/ml.
- This solution should be prepared ≤ 15 minutes prior to use and should not be reused.
Proteinase K (20 mg/ml)
- Dissolve 200 mg of proteinase K powder in 10 ml of sterile ddH2O.
- Aliquot 1 ml into microcentrifuge tubes.
- Store at -20°C.
Hyaluronidase (30 mg/ml)
- Dilute 100 mg in 3.3 ml sterile ddH2O to make 30 mg/ml solution.
- Dispense in 500 µl aliquots; store at -20°C.
Sodium acetate (3.0 M, pH 5.5)
- Dissolve 40.8 g sodium acetate in 80 ml sterile ddH2O.
- Adjust pH to 5.5 with glacial acetic acid.
- Add sterile ddH2O to final volume of 100 ml.
- Autoclave at 121°C for 20 minutes.
- Store at room temperature.
Phenol: chloroform (1:1)
- Melt solid or liquified phenol in a 68°C water bath. Liquified phenol should be stored at -20°C.
- Mix equal volumes of phenol and chloroform.
- Add an equal volume of 0.1 M Tris-HCl pH 7.6 to the phenol.
- Mix for 15 minutes and place the bottle back to water bath to allow the phases to separate.
- Remove the top aqueous layer as much as possible.
- Repeat step b-d until the top aqueous layer reaches ~pH 7.6 (pH should be measured with pH paper. Do not use a pH meter.).
- After the phenol is equilibrated, add an equal volume of 0.01 M Tris-HCl (pH 7.6).
- Store in a dark glass bottle at 4°C up to 6 months.
- Fast preparation of DNA template from clinical isolates
N. meningitidis and H. influenzae (gram-negative)
- Dispense 1.0 ml of 10 mM Tris (pH 8.0) buffer into 1.5 ml microcentrifuge tubes and label.
- Harvest colonies from 18-24 hour pure cultures of H. influenzae or N. meningitidis using a sterile polyester or rayon-tipped swab and swirl the swab in the Tris buffer to make a turbid suspension (equivalent to a McFarland 3.0 standard). Be careful not to pick up pieces of agar on the swab.
- Vortex briefly and boil cell suspension at 100°C for 10 minutes.
- Proceed immediately with PCR or store at -20°C.
Fast DNA extraction protocol for S. pneumoniae (gram-positive)
- Dispense 300 µl of 0.85% NaCl into 1.5 ml microcentrifuge tubes and label.
- Harvest colonies (use 1 loopful of a 10 µl loop) from 18-24 hour pure cultures of S. pneumoniae using a sterile polyester or rayon-tipped swab and swirl the swab in the 0.85% NaCl to make a turbid suspension (equivalent to McFarland 3.0 standard). Be careful not to pick up pieces of agar on swab.
- Vortex briefly and incubate at 70°C for 15 minutes.
- Microcentrifuge at 12,000 x g for 2 minutes and remove the supernatant.
- Re-suspend in 50 µl TE buffer (10 mM Tris-HCl, 100 µM EDTA, pH 8.0) and add 10 µl mutanolysin (3000 U/ ml)* and 8 µl of hyaluronidase (30 mg/ml)**
- Incubate at 37°C for 30 minutes up to 18 hours (overnight).
- Heat-inactivate the enzymes in the suspension by boiling at 100°C for 10 minutes.
- Microcentrifuge at 12,000 x g for 4 minutes and remove supernatant for use as DNA template.
- Proceed immediately with PCR or store at -20°C.
Footnotes
*Mutanolysin (10,000 U). Dilute in 3.3 ml of TE buffer to make 3000 U/ml stock solution, store at -20°C as 500 µl aliquots.
**Hyaluronidase (100 mg). Dilute in 3.3 ml of TE buffer to make 30 mg/ml solution, store at -20°C as 500 µl aliquots.
- Extracting genomic DNA from clinical isolates and specimens
Efficient extraction of the DNA template is a necessary step for any real-time PCR assay. The goal of DNA extraction is to lyse the bacterial cells in the specimens to maximize bacterial DNA yield and quality while removing any PCR inhibitors (i.e. salts, proteins), dissolve the DNA in a buffer compatible with the enzymes used in the next step and concentrating the DNA at the same time. When considering a DNA extraction method, it is important to select one that will produce an adequate DNA yield for detection by real-time PCR (dependant on the assay-specific lower limit of detection) without purifying potential PCR inhibitors as well. Things to consider are the type and volume of specimen, nucleic acid sought (DNA or RNA), concentration of the target DNA present in the specimen, impurities present that could act as PCR inhibitors, facilities/equipment available, and safety requirements. Generally, methods with fewer steps decrease chances of contamination and loss of DNA. Commercial methods are available for both cell lysis and purification and include silica membrane, spin column, and magnetic bead technology, in addition to biochemical and physical methods. In general, these methods produce adequate results as long as the protocol provided by the manufacturer is precisely followed.
- Bacterial cell lysis
The first step in extracting and purifying bacterial DNA is to lyse the bacterial cell walls for maximum DNA yield. There are multiple ways to lyse bacterial cells, either physically or chemically, and this step can be optimized by considering the suspected bacteria and starting specimen material, as well as the materials available to each laboratory. Chemical or enzymatic based lysis methods are typically simpler to perform and can be more cost efficient. Both N. meningitidis and H. influenzae are Gram negative and can be effectively lysed using lysis buffer containing protease such as Proteinase K along with a detergent. Incubation temperature and duration vary between organisms and specimen material. The optimal temperature range for Proteinase K activity is between 55-65°C. At temperatures above 65°C, the enzyme activity decreases. However, specimens incubated at 37°C can be left for longer incubation periods without affecting DNA quality. Specimens should be incubated until cells are completely lysed (when solution clears) and the time will vary between specimens. Once the bacteria are completely lysed one should proceed to the next step.
For optimal yields of S. pneumoniae, which is gram-positive, additional enzyme digestion with lysozyme and mutanolysin will help to degrade the higher content of peptidoglycan in the cell wall before being lysed with buffer. The temperature and length of the enzyme incubation will depend on the concentration and type of enzyme used, as well as the lysis buffer used. High temperature incubation and repeated freeze/thaw cycles are generally used with higher concentrations of cells, such as when extracting from cultures. Physical lysis can be performed using a liquid or pressure cell homogenizer, sonication, or shaking with glass beads, although some of these methods will require additional and sometimes costly equipment and they tend to shear the DNA in to smaller fragments.
Enzyme lysis for clinical specimens of unknown etiology or known gram-positive cell suspensions (S. pneumoniae)
- Prepare digestion buffer (0.04 g/ml lysozyme and 75 U/ml mutanolysin in TE buffer).
- This solution should be prepared ≤ 15 minutes before use and not reused.
- Add 100 µl of digestion buffer to each microcentrifuge tube.
- Add 200 µl of bacterial cell suspension or clinical specimen to each microcentrifuge tube. Vortex and incubate at 37°C for 1 hour.
- If specimen volume is less than 200 µl, note the volume in lab records and add TE buffer to a total volume of 200 µl. If the specimen tube appears to be empty, wash the sides of the tube with 200 µl TE buffer.
- Add 200 µl of cell lysis buffer to each microcentrifuge tube.
- Add 20 µl of Proteinase K (20 mg/ml) and invert each tube until the phases are completely mixed.
- Incubate at 37°C for 30 minutes to 1 hour.
- Purify DNA before using in real-time PCR reactions.
Enzyme lysis for known gram-negative clinical specimens or cell suspensions (N. meningitidis and H. influenzae)
- Prepare cell lysis buffer (4% SDS, 1 mM EDTA pH 8.0).
- Add 200 µl of lysis buffer to each microcentrifuge tube.
- Add 200 µl of bacterial cell suspension or clinical specimen (CSF, serum, or blood) to each microcentrifuge tube.
- Add 20 µl of Proteinase K (20 mg/ml) for a final concentration of 1 mg/ml and invert each tube until the phases are completely mixed.
- Incubate at 37°C for 30 minutes to 1 hour.
- Purify DNA before using in real-time PCR reactions.
- Prepare digestion buffer (0.04 g/ml lysozyme and 75 U/ml mutanolysin in TE buffer).
- Removal of eukaryotes from blood
If DNA is being extracted from clinical specimens such as blood, CSF, or serum, considerations should be taken to remove potential PCR inhibitors from the surrounding material before lysing the cells. If the starting specimen is blood, steps to remove erythrocytes can results in higher DNA yield with fewer PCR inhibitors. Hemoglobin is very inhibitory to DNA polymerases. Erythrocytes can be removed through the use of a hypotonic buffer or by using gradient centrifugation to create a buffy coat in which leukocytes are concentrated. Bacteria in large volumes of clinical specimens can be concentrated by centrifugation or antigen capture to increase yield.
Preparation of buffy coat
- Centrifuge the blood specimen at 2,500 x g for 10 minutes at room temperature.
- Three layers should be apparent after centrifugation. The top layer should be clear and contains plasma. The light tan middle layer is the buffy coat and contains concentrated leukocytes. The bottom red layer contains erythrocytes.
- Purification of DNA
Purification of the extraction product is important to remove any residual material that could potentially inhibit real-time PCR. Purification can be performed by many commercially available extraction kits or with the use of organic solvents, such as the chloroform/phenol method. Some methods may purify RNA along with DNA and as RNA may inhibit some reactions, use of RNAase improves purity of DNA as well.
Phenol/Chloroform to remove cell debris and proteins
Phenol is a hazardous organic solvent and safety precautions should be taken when working with phenol. Always use suitable chemical protection gloves when handling phenol containing solutions. Specific waste procedures may be required for the disposal of solutions containing phenol.
- To a lysed specimen, add an equal volume of phenol: chloroform solution (1:1). Mix well by inversion or briefly vortex.
- Centrifuge the tube at 16,000 x gfor 15 minutes in a microcentrifuge.
- Carefully remove the top aqueous layer from the bottom phenol layer and transfer to a new tube, being careful to avoid the interface.
- Steps 1-3 can be repeated until an interface is no longer visible.
- To remove all traces of phenol, add an equal volume of chloroform to the aqueous layer and centrifuge the tube at 16,000 x gfor 15 minutes in a microcentrifuge.
- Carefully remove the top aqueous layer from the bottom chloroform layer and transfer to a new tube, being careful to avoid the interface.
- Steps 5-6 can be repeated until an interface is no longer visible.
- Precipitate the DNA by ethanol or isopropanol.
Precipitation of DNA by ethanol or isopropanol
- Add a 0.1 (1/10th) volume of 3.0 M sodium acetate (pH 5.5) to the aqueous phase and then 2 volumes of 95% ethanol. Incubate at -20°C overnight or for shorter periods at -80°C (e.g. 20-30 minutes). Proceed with step 3.
- If isopropanol is used: Add a 0.1 volume of 3.0 M sodium acetate (pH 5.5) to the aqueous phase and then 0.6 volumes of 100% isopropanol. Incubate at -20°C for 2 hours or for shorter periods at -80°C (e.g., 10-20 minutes).
- Centrifuge at 16,000 x g for 30 min at 4°C.
- Recover the precipitated DNA by centrifuging the tube at 16,000 x g for 15 minutes at 4°C. Remove the aqueous phase with care.
- Add 2 volumes (of original sample) of 75% (v/v) ethanol and leave at room temperature for 5-10 minutes to remove excess salt and traces of phenol and chloroform from the pellet.
- Centrifuge at 16,000 x g for 5 minutes. Remove with care as much ethanol as possible from the microcentrifuge tube using a filtered pipette tip to avoid dislodging the pellet.
- Dry the DNA pellet in air, in a desiccator, or in a 50°C oven for 5 minutes.
- The dried DNA may be dissolved in sterile Tris buffer (10mM Tris-HCl, pH 8.0) and stored at 4°C for further manipulation or at -20°C for long-term storage.
Storage of DNA
Extracted and purified DNA should be stored in a designated elution buffer from a commercial kit or in Tris buffer (10 mM Tris-HCl, pH 8.0). Distilled water can also be used but these specimens may experience degradation from acid hydrolysis. DNA can be kept at 4°C for short periods of time and at -20°C for long-term storage.
- Bacterial cell lysis
- Alternative protocol for genomic DNA extraction: Boom method
Reagent preparation
Extraction buffer L6
- Add 120 g of guanidinium isothiocyanate (GuSCN) to 100 ml of 0.1 M Tris/HCl (pH 6.4) and 22 ml of 0.2 M EDTA (pH 8.0) and 2.6 g of Triton X-100.
- Stir overnight in the dark to dissolve.
- Store away from light for up to 1 month.
- GuSCN is toxic and care should be taken when handling this substance.
Extraction buffer L2
- Add 120 g of guanidinium isothiocyanate (GuSCN) to 100 ml of 0.1 M Tris/HCl (pH 6.4).
- Stir overnight in the dark to dissolve.
- Store away from light for 1 month.
Size fractionated silica
- Add 60 g of silicon dioxide to 500 ml of distilled water in a graduated cylinder and leave at room temperature for 24 hours.
- Remove and discard 430 ml of supernatant and re-suspend solids in 500 ml of ddH2O.
- Leave at room temperature for 5 hours and remove and discard 440 ml of supernatant.
- Add 600 µl of concentrated HCl (pH 2.0), mix, and aliquot into 1.5 ml volumes.
- Sterilize by autoclaving and store away from light for up to 6 months.
DNA isolation
- Add 100 µl of specimen (bacterial suspension or clinical specimen) to 500 µl of L6 extraction buffer and 10 µl of size fractionated silica in a 1.5 ml microcentrifuge tube. For double volumes of specimen, double the amount of L6 extraction buffer and silica, as well as reagents in the wash steps (L2 extraction buffer, ethanol and acetone).
- Vortex the tube for 10 seconds and incubate, with shaking, at room temperature for 15 minutes.
- Centrifuge the tube for 15 seconds at 16,100 x g and dispose of the supernatant.
- Wash the pellet two times with 500 µl L2 extraction buffer, two times with 500 µl of 70% ethanol and one time with 500 µl of acetone. Centrifuge for 15 seconds at 16,100 x g after each wash and dispose of the supernatant following appropriate procedures for chemical waste.
- To remove the acetone, place the tube with the lid open at 56°C in a dry heating block for 5 minutes.
- Elute the nucleic acid from the silica by adding 30 µl of distilled water, close the tube, vortex, and incubate at 56°C for 15 minutes.
- Centrifuge the tube at 16,100 x g for 2 minutes and collect the supernatant, taking care not to include any silica. The extracted DNA can be stored at 4°C overnight or at -70°C for long-term storage.
- General considerations
- Conventional PCR
- General considerations
PCR is a very sensitive method for amplifying a specific DNA target, but also very susceptible to contamination with extraneous DNA. Extra precautions should be taken to minimize such cross-contamination. It is recommended to physically separate the different steps including PCR reaction assembly, addition of template DNA to the reaction wells, and agarose gel detection of PCR products. If separate rooms are not possible, separate laboratory benches should be used for these steps. Working in an unventilated still air biocontainment cabinet (sometimes called a PCR hood), are also suggested to minimize cross-contamination. Separate micropipettors, laboratory coats, and gloves should be used for reaction assembly. Signage indicating which spaces and equipment are DNA-free and which are used to prepare or manipulate DNA would be helpful. Decontamination of surfaces and equipment with 10% bleach followed by 70% ethanol should be performed after any manipulation of nucleic acids at the laboratory bench. Always use filter-barrier pipette tips and change and discard gloves frequently.
- Equipment, consumables, and reagents
Equipment:
- PCR thermocycler
- Unventilated biocontainment cabinet or PCR hood
- Freezer
- Refrigerator
- Electrophoresis tank
- Power supply
- Stir plate
- Microwave oven
- Gel viewing system
- Gel documentation system
Consumables:
- 10% bleach (10:1, water: concentrated bleach) (make fresh weekly)
- 70% ethanol
- 1.5 mL microcentrifuge tubes (sterile DNase free or PCR grade)
- 96 well polypropylene plates, tube strips or individual
- 1 set of micropipettors (1-10 µl, 2-20 µl, 20-200 µl, and 100-1000 µl)
- Pre-sterilized filter tips (10 µl, 200 µl, and 1000 µl)
- Optical caps
Optional:
- Commercial DNA-removing surface decontaminant liquid
- Cap installing tool
Reagents:
- DNA polymerase
- dNTPs
- Primers Positive and negative control DNA diluted to approximately 5 µg/ml
- PCR grade water
- TAE or TBE buffer
- Agarose powder (molecular biology grade)
- DNA ladder
- 6x DNA loading dye
- Ethidium bromide
- Bromophenol blue
- Xylene cyanol FF
- Sucrose
- Preparing reagent stock solution and primer working solution
EDTA, 0.5 M, pH 8.0 (100 ml)
- Dissolve 18.6 g EDTA in 70 ml ddH2O.
- Adjust pH to 8.0 with 10 M NaOH (~5 ml).
- Add ddH2O to 100 ml and mix well on a stir plate.
- Store at room temperature.
Ethidium bromide (EtBr), 10 mg/ml
- Dissolve 0.2 g ethidium bromide in 20 ml ddH2O.
- Mix well and store at 4°C in the dark in 1 ml aliquots.
- Store at room temperature.
TAE (Tris/acetate/EDTA) electrophoresis buffer, 50 X stock solution*
- To 750 ml of ddH2O add:
- 242 g Tris base
- 57.1 ml of glacial acetic acid
- 100 ml of 0.5 M EDTA pH 8.0
- Add ddH2O to 1000 ml and mix well on a stir plate.
- Store at room temperature.
Footnote
*TAE stock solution should be diluted to 1X in H2O before use.
TBE (Tris/borate/EDTA) electrophoresis buffer, 10X stock solution*
- To 900 ml of ddH2O add:
- 108 g Tris base (890 mM)
- 55 g boric acid (890 mM)
- 40 ml 0.5 M EDTA, pH 8.0 (20 mM)
- Add ddH2O to 1000 ml and mix well on a stir plate.
- Store at room temperature.
Footnote
*TBE stock solution should be diluted to 0.5X in H2O before use.
6X DNA loading dye I
- 0.25% bromophenol blue
- 0.25% xylene cyanol FF
- 40% (w/v) sucrose in water
- Store at 4°C.
6X DNA loading dye II
- 0.25% bromophenol blue
- 0.25% xylene cyanol FF
- 30% glycerol in water
- Store at 4°C.
6X DNA loading dye III
- 0.25% bromophenol blue
- 40% (w/v) sucrose in water
- Store at 4°C.
2% agarose gel
- Add 2 g of electrophoresis-grade agarose to 100 ml of 1X TAE or 0.5X TBE buffer in a 250 ml flask or bottle.
- Melt the agarose in a microwave until the agarose is fully melted and the solution is clear. Swirl the flask a few times while microwaving to avoid boiling and spilling over.
- Cool to 55-60°C and then add 5 µl EtBr for a final concentration of 0.5 µg/ml.
- EtBr is a powerful carcinogen and must be handled with care.
Primer working stock solution
The primer working stock solution should be 20 µM.
- Performing multiplex PCR for S. pneumoniae serotype deduction
PCR protocol
Prior to beginning the PCR, plan the experiment by filling out and printing a plate template worksheet. Also, be sure sufficient quantities of primer working solutions to be used are available.
- Remove DNA templates and positive control DNAs from -20°C to the DNA addition area to allow them to thaw completely.
- In the PCR reaction assembly area, gather reagents needed for the PCR reactions, including PCR master mix, primers, and PCR grade water. If the reagents are stored at -20°C, allow them to thaw completely and vortex or flick each tube before use.
- Sequential multiplex PCR reactions, based on one of the described schemes, are prepared in standard 25 µl reaction volumes using primers and concentrations described in Table 5.
- Each PCR reaction should contain:
- A master mix can be prepared, which includes all components listed above except DNA template. When calculating volumes of master mix reagents, remember to add enough master mix reagents for 2 extra reactions than the number of specimens there are to be tested to ensure there will be enough master mix.
- Pipette 22.5 µl of this master mix into each appropriate well of 96-well plate, according to your plate template worksheet.
- Cover the wells of the plate using cap strips. Spray down the clean workspace with 10% bleach (10:1 water: concentrated bleach), and wipe. Repeat with 70% alcohol. Remove laboratory coat and gloves. Put on a fresh pair of gloves. Carefully transport the 96-well plate to the DNA addition area.
- Put on new laboratory coat and keep the same pair of gloves on. Remove the cap strips from the plate. Add 2.5 µl of template DNA to each appropriate well of 96-well plate, according to your plate template worksheet.
- At least one negative and one positive control should be set up for each serotype per PCR run.
- Negative control: add 2.5 µl DNA resolving buffer to a reaction well instead of DNA template.
- Positive control: add 2.5 µl of DNA template that is known to contain the amplified sequence to a reaction well.
- Cap columns of wells as you go. Use the roller tool to secure caps tightly.
- Wipe down the dirty workspace with 10% bleach, then 70% ethanol. Remove laboratory coat and gloves. If possible, quickly spin the plate at 1000 rpm to bring down any droplets. Transport plate directly to and place it in the PCR thermocycler.
- The following PCR conditions are used:
- 1 cycle of 94°C for 4 minutes
- 30 cycles of 94°C for 45 seconds; 54°C for 45 seconds and 72°C for 2 minutes
- 1 cycle of 72°C for 2 minutes
- Current methodologies are detailed at the CDC website
Analysis of PCR products on an agarose gel
PCR products (10 µl) are run on 2% agarose gels to determine band sizes using positive controls. A positive control for each serotype and a 50 bp ladder molecular size marker should be included on each gel.
- Melt the 2% agarose gel in a microwave oven. Cool the agar to approximately 55°C. Add ethidium bromide or other gel stain. Pour into a gel casting cassette, insert the comb, and allow time for hardening (~30 minutes).
- Add 1X TAE or TBE buffer to the electrophoresis tank and properly place the gel cassette containing the solidified agarose gel into the tank.
- Briefly spin the PCR plate or tubes at 500 x g to ensure all liquid is at the bottom.
- Mix 10 µl of PCR reaction with 2 µl of 6X loading dye.
- Pipette the DNA/loading dye mixtures into the wells. Load 5 µl of DNA size markers in one of the wells.
- Run the gel at 50-100 volts for 15-20 minutes or until the Bromophenol blue dye band is halfway down the gel. The dye runs at approximately the same rate as a 500 base-pair DNA fragment.
- Visualize the gel under a UV light and print out or save the image, if possible.
- Each reaction should give two bands, i.e., species-specific positive control (cpsA, although some are cpsA negative) and a serotype-specific band.
- Store the remainder of the amplicon at -20°C, if necessary.
Interpretation
Band sizes on agarose gelsmust match those of positive controls before assigning a putative serotype (Figure 5). PCR reactions are setup sequentially and will include those serotypes most frequently determined for each of the schemes. For example, if a strain is negative for any of the serotypes included in reaction 1 then proceed to reaction 2 and so forth until a serotype is determined. If all reactions are completed for a specific scheme and no serotype bands were identified that matched any of the positive controls then the strain may be a nontypeable or one of the serotypes not yet included in the scheme and would need to be further typed using the Quellung reaction (see Chapter 8: Identification and Characterization of Streptococcus pneumoniae). Resolution of individual serotypes within some of the positive reactions could be applicable in some circumstances. For example, to further resolve a PCR positive reaction for 12F/12A/44/46 (reaction 3 in Table 6), perform a Quellung reaction using type-specific antisera to determine whether the strain has a 12F, 12A, 44, or 46 capsular type. It must be mentioned that for practical reasons this is usually not necessary. For example, serotype 12F is commonly detected in disease and carriage specimens, while serotypes 12A, 44, and 46 are extremely rare.
Helpful tips
The positive pneumococcal control band for cpsA can be negative in 1-2% of PCR-serotypeable isolates. This is most often encountered in serotypes 25 and 38, but has also rarely been found for serotypes 14 and 35A. Although a cpsA negative result is relatively rare, at present a negative cpsA does not necessarily equate to a non-serotypeable isolate or a pneumococcus-negative clinical specimen.
Quality control
A positive control for each serotype included in the PCR reaction(s) should be run on each gel to ensure that bands are assigned to the correct serotype.
Table 2. Primers and probes used for detection of bacterial meningitis pathogens
Target Primer or Probe Name Real-time Primers and Probes Nucleotide Sequence (5' to 3') Working Stock Conc (µM) Final Conc (nM) Suggested Probe Modifications N. meningitidis ctrA F753 TGTGTTCCGCTATACGCCATT 3.75 300 R846 GCCATATTCACACGATATACC 11.25 900 Pb820i AACCTTGAGCAA"T"CCATTTA
TCCTGACGTTCT1.25 100 5' FAM, BHQ1 on "T", 3' SpC6 sodC F351 GCACACTTAGGTGATT
TACCTGCAT3.75 300 R478 CCACCCGTGTGGATCATAATAGA 7.5 600 Pb387 CATGATGGCACAGCAACAAA
TCCTGTTT1.25 100 5' FAM, 3' BHQ1 H. influenzae hpd hpdF822 GGTTAAATATGCCGATGGTGTTG 1.25 100 hpdR952 TGCATCTTTACGCACGGTGTA 3.75 300 Pb896i TTGTGTACACTCCGT"T"GGT
AAAAGAACTTGCAC1.25 100 5' FAM, BHQ1 on "T", 3' SpC6 S. pneumoniae lytA F373 ACGCAATCTAGCAGATGAAGCA 2.5 200 R424 TCGTGCGTTTTAATTCCAGCT 2.5 200 Pb400i TGCCGAAAACGC”T”TGATAC
AGGGAG2.5 200 5' FAM, BHQ1 on "T", 3' SpC6 Human DNA RNAaseP RNAsePF CCA AGT GTG AGG GCT GAA AAG 4 400 RNAsePR TGT TGT GGC TGA TGA
ACT ATA AAA GG4 400 RNAsePPb CCC CAG TCT CTG TCA GCA
CTC CCT TC1 100 5' FAM, 3' BHQ1 Table 3. Primers and probes used for detection of the serogroups of N. meningitidis
Target Primer or Probe Name Real-time Primers and Probes Nucleotide Sequence (5' to 3') Working Stock Conc (µM) Final Conc (nM) Suggested Probe Modifications Nm A F2531 AAAATTCAATGGGTATATCACGAAGA 3.75 300 sacB R2624 ATATGGTGCAAGCTGGTTTCAATAG 11.25 900 Pb2591i CTAAAAG"T"AGGAAGGGCACT
TTGTGGCATAAT1.25 100 5' FAM, BHQ1 on "T", 3' SpC6 Nm B F737 GCTACCCCATTTCAGATGATTTGT 3.75 300 synD R882 ACCAGCCGAGGGTTTATTTCTAC 3.75 300 Pb839i AAGAGATGGGYAACAAC"T"ATGT
AATGTCTTTATTT1.25 100 5' FAM, BHQ1 on "T", 3' SpC6 Nm C F478 CCCTGAGTATGCGAAAAAAATT 11.25 900 synE R551 TGCTAATCCCGCCTGAATG 3.75 300 Pb495i TTTCAATGC"T"AATGAATACCAC
CGTTTTTTTGC1.25 100 5' FAM, BHQ1 on "T", 3' SpC6 Nm W135 F857 TATTTATGGAAGGCATGGTGTATG 1.25 100 synG R964 TTGCCATTCCAGAAATATCACC 11.25 900 Pb907i AAATATGGAGCGAATGATTACAGT
AACTATAATGAA2.5 200 5' FAM, BHQ1 on "T", 3' SpC6 Nm X F173 TGTCCCCAACCGTTTATTGG 11.25 900 xcbB R237 TGCTGCTATCATAGCCGCC 11.25 900 Pb196 TGTTTGCCCACATGAATGGCGG 1.25 100 5' FAM, 3' BHQ1 Nm Y F787 TCCGAGCAGGAAATTTATGAGAATAC 11.25 900 synF R929 TTGCTAAAATCATTCGCTCCATAT 7.5 600 Pb1099i TATGGTG"T"ACGATATCCCTATCC
TTGCCTATAAT1.25 100 5' FAM, BHQ1 on "T", 3' SpC6 Table 4. Primers and probes used for detection of the serotypes of H. influenzae
Target Primer or Probe Name Real-time Primers and Probes Nucleotide Sequence (5' to 3') Working Stock Conc (µM) Final Conc (nM) Suggested Probe Modifications Hi a F261 GGT CTG CGG TGT CCT GTG T 1.25 100 acsB R427 CCG GTC ATC TTT TAT GCT CCA A 3.75 300 Pb375i TAA TTT TCT TGC "T"CA ATA CCG CCT TCC CA 1.25 100 5' FAM, BHQ1 on "T", 3' SpC6 Hi b F192 TGA TGC ATT GAA AGA AGG TGT AAT TT 3.75 300 bcsB R359 CCT GCG GTA ATA ACA TGA TCA TAA A 7.5 600 Pb244i TGT CGT GCA G”T”A GCA AAC CGT AAC CTT ACT C 1.25 100 5' FAM, BHQ1 on "T", 3' SpC6 Hi c F7667 CAT TGG TGA TGG TTC AGT TAT TGG 7.5 600 ccsD R7784 TAC AGC ATT CAG CAA TAA TGG G 3.75 300 Pb7726i ATT GCA “T”CG CCG CAG GAG TTC CCG 2.5 200 5' FAM, BHQ1 on "T", 3' SpC6 Hi d F2211 CCT AAA ATA CGG ACC TAG TGC AC 7.5 600 dcsE R2255 CCG ATG AGA CCA AGT ATG GTT A 1.25 100 Pb2221i AAC GAG C”T”A GAG CTG GTG CTG AA 3.75 300 5' FAM, BHQ1 on "T", 3' SpC6 Hi e F1523 ACT AAA ATA TGG CCC AAA CCC AC 7.5 600 ecsH R1589 CCG ATG AGC CCA AGT ATG ATG A 7.5 600 Pb1555i AAC GAG CAA AAG CCG G"T"G CGG AT 2.5 200 5' FAM, BHQ1 on "T", 3' SpC6 Hi f F7164 CCC TGA AAA GCG TTG ACT TTG 7.5 600 bexD R7313 CCA ACT TCA GGA CCA AGT CAT TC 3.75 300 Pb7242i TGC TGC TAA C"T"C AGA TGC ATC AGC TCC TT 2.5 200 5' FAM, BHQ1 on "T", 3' SpC6 Table 5. List of primers used for pneumococcal serotype deduction. Please note that this website should be used as the primary source of primer sequences and protocols due to periodic introduced improvements.
*All serotypes that are co-detected are listed
Target Primer or Probe Name Real-time Primers and Probes Nucleotide Sequence (5' to 3') Working Stock Conc (µM) Final Conc (nM) 1-f CTC TAT AGA ATG GAG TAT ATA AAC TAT GGT TA wzy 0.3 280 1-r CCA AAG AAA ATA CTA ACA TTA TCA CAA TAT TGG C 0.3 2-f TAT CCC AGT TCA ATA TTT CTC CAC TAC ACC wzy 0.3 290 2-r ACA CAA AAT ATA GGC AGA GAG AGA CTA CT 0.3 3-f ATG GTG TGA TTT CTC CTA GAT TGG AAA GTA G galU 0.3 371 3-r CTT CTC CAA TTG CTT ACC AAG TGC AAT AAC G 0.3 4-f a CTG TTA CTT GTT CTG GAC TCT CGA TAA TTG G wzy 0.3 430 4-r GCC CAC TCC TGT TAA AAT CCT ACC CGC ATT G 0.3 5-f ATA CCT ACA CAA CTT CTG ATT ATG CCT TTG TG wzy 0.3 362 5-r GCT CGA TAA ACA TAA TCA ATA TTT GAA AAA GTA TG 0.3 6A/6B/6C/6D -f AAT TTG TAT TTT ATT CAT GCC TAT ATC TGG wciP 0.3 250 6A/6B/6C/6D -r TTA GCG GAG ATA ATT TAA AAT GAT GAC TA 0.3 6C/6D -f CAT TTT AGT GAA GTT GGC GGT GGA GTT wciNbeta 0.5 727 6C/6D -r AGC TTC GAA GCC CAT ACT CTT CAA TTA 0.5 7C/(7B/40)-f CTA TCT CAG TCA TCT ATT GTT AAA GTT TAC GAC GGG A wcwL 0.3 260 7C/(7B/40)-r GAA CAT AGA TGT TGA GAC ATC TTT TGT AAT TTC 0.3 7F/7A-f TCC AAA CTA TTA CAG TGG GAA TTA CGG wzy 0.4 599 7F/7A-r ATA GGA ATT GAG ATT GCC AAA GCG AC 0.4 8-f GAA GAA ACG AAA CTG TCA GAG CAT TTA CAT wzy 0.2 201 8-r CTA TAG ATA CTA GTA GAG CTG TTC TAG TCT 0.2 9N/9L-f GAA CTG AAT AAG TCA GAT TTA ATC AGC wzx 0.5 516 9N/9L-r ACC AAG ATC TGA CGG GCT AAT CAA T 0.5 9V/9A-f GGG TTC AAA G TC AGA CAG TG A ATC TTA A wzy 0.5 816 9V/9A-r CCA TGA ATG A AA TCA ACA TT G TCA GTA GC 0.5 10A-f GGT GTA GAT TTA CCA TTA GTG TCG GCA GAC wcrG 0.5 628 10A-r GAA TTT CTT CTT TAA GAT TCG GAT ATT TCT C 0.5 10F/(10C/33C)- f GGA GTT TAT CGG TAG TGC TCA TTT TAG CA wzx 0.3 248 10F/(10C/33C)-r CTA ACA AAT TCG CAA CAC GAG GCA ACA 0.3 11A/11D-f GGA CAT GTT CAG GTG ATT TCC CAA TAT AGT G wzy 0.3 463 11A/11D-r GAT TAT GAG TGT AAT TTA TTC CAA CTT CTC CC 0.3 12F/(12A/44/46)-f GCA ACA AAC GGC GTG AAA GTA GTT G wzx 0.5 376 12F/(12A/44/46)-r CAA GAT GAA TAT CAC TAC CAA TAA CAA AAC 0.5 13-f TAC TAA GGT AAT CTC TGG AAA TCG AAA GG wzx 0.4 655 13-r CTC ATG CAT TTT ATT AAC CG C TTT TTG TTC 0.4 14-f GAA ATG TTA CTT GGC GCA GGT GTC AGA ATT wzy 0.3 189 14-r GCC AAT ACT TCT TAG TCT CTC AGA TGA AT 0.3 15A/15F-f ATT AGT ACA GCT GCT GGA ATA TCT CTT C wzy 0.3 434 15A/15F-r GAT CTA GTG AAC GTA CTA TTC CAA AC 0.3 15B/15C-f TTG GAA TTT TTT AAT TAG TGG CTT ACC TA wzy 0.3 496 15B/15C-r CAT CCG CTT ATT AAT TGA AGT AAT CTG AAC C 0.3 16F-f, GAA TTT TTC AGG CGT GGG TGT TAA AAG wzy 0.4 717 16F-r CAG CAT ATA GCA CCG CTA AGC AAA TA 0.4 17F-f TTC GTG ATG ATA ATT CCA ATG ATC AAA CAA GAG wciP 0.5 693 17F-r GAT GTA ACA AAT TTG TAG CGA CTA AGG TCT GC 0.5 18/(18A/18B/18C/18F)-f CTT AAT AGC TCT CAT TAT TCT TTT TTT AAG CC wzy 0.3 573 18/(18A/18B/18C/18F)-r TTA TCT GTA AAC CAT ATC AGC ATC TGA AAC 0.3 19A-f GAG AGA TTC ATA ATC TTG CAC TTA GCC A wzy 0.3 566 19A-r CAT AAT AGC TAC AAA TGA CTC ATC GCC 0.3 19F-f GTT AAG ATT GCT GAT CGA TTA ATT GAT ATC C wzy 0.5 304 19F-r GTA ATA TGT CTT TAG GGC GTT TAT GGC GAT AG 0.5 20-f GAG CAA GAG TTT TTC ACC TGA CAG CGA GAA G wciL 0.3 514 20-r CTA AAT TCC TGT AAT TTA GCT AAA ACT CTT ATC 0.3 21-f CTA TGG TTA TTT CAA CTC AAT CGT CAC C wzx 0.2 192 21-r GGC AAA CTC AGA CAT AGT ATA GCA TAG 0.2 22F/22A-f GAG TAT AGC CAG ATT ATG GCA GTT TTA TTG TC wcwV 0.5 643 22F/22A-r CTC CAG CAC TTG CGC TGG AAA CAA CAG ACA AC 0.5 23A-f TAT TCT AGC AAG TGA CGA AGA TGC G wzy 0.5 722 23A-r CCA ACA TGC TTA AAA ACG CTG CTT TAC 0.5 23B-f CCA CAA TTA G CG CTA TAT TCA TTC AAT CG wzx 0.2 199 23B-r GTC CAC GCT GAA TAA AAT GAA GCT CCG 0.2 23F-f a GTA ACA GTT GCT GTA GAG GGA ATT GGC TTT TC wzy 0.5 384 23F-r CAC AAC ACC TAA CAC TCG ATG GCT ATA TGA TTC 0.5 24A/24B/24F-f GCT CCC TGC TAT TGT AAT CTT TAA AGA G wzy 0.2 99 24A/24B/24F-r GTG TCT TTT ATT GAC TTT ATC ATA GGT CGG 0.2 31-f GGA AGT TTT CAA GGA TAT GAT AGT GGT GGT GC wzy 0.5 701 31-r CCG AAT AAT ATA TTC AAT ATA TTC CTA CTC 0.5 33F/33A/37-f GAA GGC AAT CAA TGT GAT TGT GTC GCG wzy 0.3 338 33F/33A/37-r CTT CAA AAT GAA GAT TAT AGT ACC CTT CTA C 0.3 34-f GCT TTT GTA AGA GGA GAT TAT TTT CAC CCA AC wzy 0.3 408 34-r CAA TCC GAC TAA GTC TTC AGT AAA AAA CTT TAC 0.3 35A/35C/42-f ATT ACG ACT CCT TAT GTG ACG CGC ATA wzx 0.3 280 35A/35C/42-r CCA ATC CCA AGA TAT ATG CAA CTA GGT T 0.3 35B-f GAT AAG TCT GTT GTG GAG ACT TAA AAA GAA TG wcrH 0.5 677 35B-r CTT TCC AGA TAA TTA CAG GTA TTC CTG AAG CAA G 0.5 35F/47F-f GAA CAT AGT CGC TAT TGT ATT TTA TTT AAA GCA A wzy 0.3 517 35F/47F-r GAC TAG GAG CAT TAT TCC TAG AGC GAG TAA ACC 0.3 38/25F-f CGT TCT TTT ATC TCA CTG TAT AGT ATC TTT ATG wzy 0.3 574 38/25F-r ATG TTT GAA TTA AAG CTA ACG TAA CAA TCC 0.3 39-f TCA TTG TAT TAA CCC TAT GCT TTA TTG GTG wzy 0.2 98 39-r GAG TAT CTC CAT TGT ATT GAA ATC TAC CAA 0.2 cpsA-f GCA GTA CAG CAG TTT GTT GGA CTG ACC wzg 0.1 160 cpsA-r GAA TAT TTT CAT TAT CAG TCC CAG TC 0.1 Table 6. PCR-serotyping scheme for USA based on current serotype prevalence
Reaction Serotypes included 1 6A/6B/6C/6D, 3, 19A, 22F/22A, 16F 2 8, 33F/33A/37, 15A/15F, 7F/7A, 23A 3 19F, 12F/12A/44/46, 11A/11D, 38, 35B 4 24A/24B/24F, 7C/7B/40, 4, 18A/18B/18C/18F, 9V/9A 5 14, 1, 23F, 15B/15C, 10A 6 39, 10F/10C/33C, 5, 35F/47F, 17F 7 23B, 35A/35C/42, 34, 9N/9L, 31 8* 6A/6B/6C/6D, 6C/6D *Only performed if PCR-positive for 6A/6B/6C/6D in reaction 1
Table 7. PCR-serotyping scheme for Africa based on current serotype prevalence
Reaction Serotypes included 1 14, 1, 5, 4, 18A/18B/18C/18F 2 6A/6B/6C/6D, 19F, 23F, 38/25F, 9V/9A 3 7C/7B/40, 3, 15B/15C, 7F/7A, 17F 4 8, 12F/12A/44/46, 9L/9N, 22F/22A, 23A 5 24A/24B/24F, 2, 11A/11D, 19A, 16F 6 21, 33F/33A/37, 15A/15F, 35F/47F, 13 7 39, 23B, 35A/35C/42, 20, 35B 8 10F/10C/33C, 34, 10A, 31 9* 6A/6B/6C/6D, 6C/6D *Only performed if PCR-positive for 6A/6B/6C/6D in reaction 2
Table 8. PCR-serotyping scheme for Latin America based on current serotype prevalence
Reaction Serotypes included 1 14, 6A/6B/6C/6D, 23F, 19A, 9V/9A 2 19F, 3, 15B/15C, 18A/18B/18C/18F, 17F 3 1, 5, 9L/9N, 7F/7A, 16F 4 8, 2, 4, 20, 22F/22A 5 7C/7B/40, 12F/12A/44/46, 11A/11D, 10A, 23A 6 21, 33F/33A/37, 15A/15F, 35F/47F, 13 7 39, 23B, 35A/35C/42, 38/25F/25A, 35B 8 24A/24B/24F, 10F/10C/33C, 34, 31 9* 6A/6B/6C/6D, 6C/6D *Only performed if PCR-positive for 6A/6B/6C/6D in reaction 1
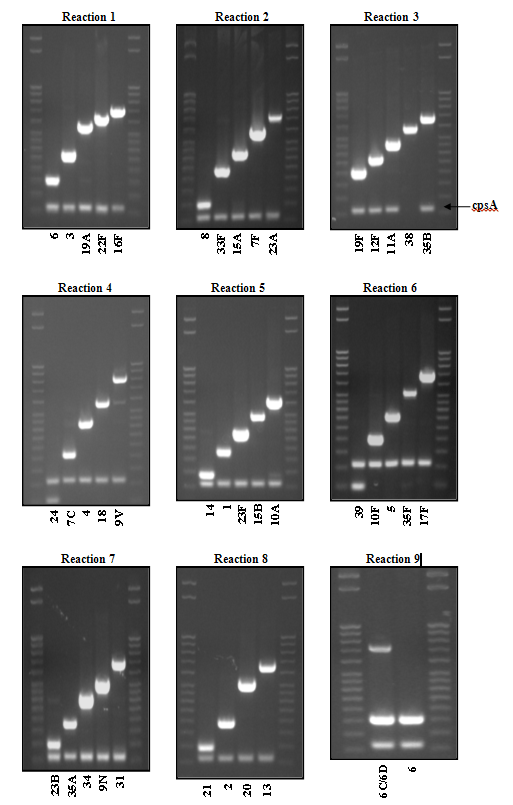
Figure 5. PCR products for USA serotyping scheme detailed in Table 6...view larger
- General considerations
- Real-time PCR
- Workstation for real-time PCR reaction set-up
The extremely sensitive lower limit of detection of real-time PCR assays increases the chance of detection of cross-contamination with other DNA. Extra precautions should be taken to minimize such cross-contamination. It is strongly recommended that reaction assembly be performed in one room, which is designated a clean room, while DNA extraction from clinical specimens or isolates and addition of template DNA to the reaction wells should be performed in a separate room, which is designated a dirty room. If separate rooms are not possible, separate laboratory benches should be used for these two steps. Working in an unventilated biocontainment cabinet (sometimes called a PCR hood), is also suggested to minimize cross-contamination. Use of a biological safety cabinet is necessary for infectious materials but does not provide significant protection from amplicon cross-contamination. Separate micropipettors, laboratory coats, and gloves should be used for DNA extraction and reaction assembly. Signage indicating which spaces and equipment are DNA-free and which are used to prepare or manipulate DNA would be helpful. Decontamination of surfaces and equipment with 10% bleach followed by 70% ethanol should be done after any manipulation of nucleic acids at the laboratory bench. Always use filter-barrier pipette tips and change and discard gloves frequently. Even though real-time PCR is more expensive, it may be advisable to use given that it is a closed system and has much less potential for cross-contamination of the workspace.
- Equipment for real-time PCR analysis
The main piece of equipment needed is the real-time PCR machine. There are more than 30 different real-time PCR thermocycler models that are manufactured by 14 companies (13). When choosing the machine to buy, be sure to consider the fluorescent filters that it will contain; the filters in the machine must be able to detect the wavelengths of light that will be emitted by the fluorophores conjugated to the probes. An accompanying desktop or laptop computer and appropriate software will be necessary to view and analyze the results that are generated by the machine. Given their replacement cost and sensitivity to damage by electrical surges, it is strongly recommended that this machine and computer be plugged directly into a battery backup with surge protection, to protect them from fluctuations in current, to protect your data, and to keep the reactions running in case power is lost.
- Consumables and reagents
Consumables:
- 10% bleach (10:1, water: concentrated bleach) (make fresh weekly) 70% ethanol
- 1.5 ml microcentrifuge tubes (sterile, DNase free, or PCR grade)
- 96 well polypropylene plates, tube strips or individual PCR tubes
- 1 set of micropipettors (1-10 µl, 2-20 µl, 20-200 µl, and 100-1000 µl)
- Pre-sterilized filter tips (10 µl, 200 µl, and 1000 µl) Optical caps
Optional:
- Optical adhesive film
- Commercial DNA-removing surface decontaminant liquid
- Cap installing tool
Reagents:
- TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA)
- Tris buffer (10 mM Tris-HCl, pH 8.0)
- Commercial PCR Master Mix (containing dNTPs, DNA polymerase, and reference dye)
- Primers and dual-labeled hydrolysis probes (probes contain 5' fluorophore and 3' or internal quencher)
- Positive control DNA diluted to about 5 µg/ml
- PCR grade water
- Preparing primer and probe working stocks
- The protocols, primers, and probes described in this chapter are adaptations of those previously described (8, 35, 60).
Table 2 lists the recommended nucleotide sequences, working concentrations, and suggested chemical modifications of the primers and probes for N. meningitidis, H. influenzae, and S. pneumoniae species detection targeting the ctrA or sodC, hpd, and lytA genes, respectively. Primers and probes for the detection of N. meningitidis serogroups (Table 3) and H. influenzae serotypes (Table 4) are also given. Additional S. pneumoniae serotype assays are currently in development. A two-tiered approach for detecting and characterizing bacterial meningitis pathogens is recommended with the first tier being species-specific detection of N. meningitidis, H. influenzae, or S. pneumoniae with subsequent serogroup/serotype determination of any positive specimens (Figure 8).
Primers and probes must be diluted from concentrated stocks into working stocks. It is convenient to dilute working stocks to concentrations that will allow 2 µl of each primer and probe to be added to the master mix per reaction. The optimized working stock concentration of each primer and probe for the assays described here that will allow 2 µl of each to be added to the master mix is presented in the tables above.
To calculate the required dilution from concentrated stock to working stocks, use the formula: (Concentration1)(Volume1)=(Concentration2)(Volume2). Example: A 500 µl working stock of the sacB forward primer is needed. For the purposes of this example, the sacB forward primer has a stock concentration of 220 µM. Table 3 shows that the working concentration of the sacB forward primer should be 3.75 µM; therefore the equation becomes:
(220 µM is the given concentration of the concentrated stock)(x µl of concentrated stock) = (3.75 µM is the desired concentration of the working stock)(500 µl is the desired volume of working stock)
Or: (220 µM)(x µl) / (3.75 µM)(500 µl)
Solve for x. x = (3.75 µM)(500 µl) / 220 µM = (1875 µM/µl) / 220 µM = 8.5 µl of concentrated stock.
Next, to calculate how much water to add to x µl of concentrated stock calculated above:
500 µl total volume of working stock - x µl of concentrated stock = y µl of water to addTherefore, using the example above: 500 µl - 8.5 µl = 491.5 µl of water.
Of this working stock of the primer, add 2 µl to the master mix per specimen to be tested.
When diluting primers and probes, it is important to use filter tips and to work in a clean space (i.e., free of template DNA) to avoid cross-contamination of these reagents. It is optimal to use a clean PCR cabinet or hood if one is available. Concentrated stocks of primers and probes should be stored at -20°C. If used on a regular basis, the working stocks can be stored at 4°C. Be sure to store probes in the dark, since they are light-sensitive (ideally, covered in aluminum foil and in a box). Concentrated stocks of probes are especially susceptible to degradation due to their fluorescent tags and should not be freeze-thawed more than 5 times. Primers should not be freeze-thawed more than 20 times (46). Therefore, it is recommended that the primer and probe stocks be aliquoted upon their arrival, before initially freezing them for longer-term storage.
The length of time for which primers and probes can be stored in this manner will vary. Performing positive controls with each reaction will help you to determine when your primers and probes need to be replaced.
- Performing real-time PCR
General considerations
Before setting up the reactions, the real-time PCR machine should be turned on. For some machines, it can take about 20 minutes for the lamp to warm up.
The high sensitivity of real-time PCR significantly increases the risk for cross-contamination. Therefore, the following precautions are recommended:
- Separate rooms are suggested for handling DNA template (dirty room) and for handling other reagents (clean room) to avoid cross-contamination. Post signs to designate these areas.
- Always use barrier pipette tips.
- Always use clean laboratory coats. Use a different laboratory coat while in each of the separate rooms. Change gloves frequently, and be sure to wear fresh gloves when moving from the clean room to the dirty room.
- Use separate pipette sets in each room.
Assays are carried out in 25 µl reaction volumes, using a commercial PCR master mix according to the manufacturer's instructions. Per sample to be tested, each reaction mix contains: 2 µl of DNA sample, 2 µl of each primer, 2 µl of probe, 12.5 µl of master mix, and 4.5 µl of sterile, PCR-grade water. Add enough reagents for 1-2 extra reactions than the number of specimens there are to be tested to ensure there will be enough mix.
Positive and negative controls are extremely important to ensure the laboratorian that contamination of reagents and workspace has not generated false positives and that the assay is detecting targets as expected. Non-template controls (NTCs) contain all reagents except for template DNA; instead, 2 µl of sterile, PCR-grade water should be added to the NTC reaction wells. It is advisable to add water used in the clean room to at least two NTC reaction wells and to add water used in the dirty room to at least two NTC reaction wells. If either NTC reaction generates an amplification curve that crosses the threshold, the water from that room should be discarded and replaced, and NTCs should be performed again to determine if other reagents are the source of contamination. An additional control for contamination at the DNA extraction step is the extraction of water, as suggested in the extraction protocol. If the extracted water negative control(s) generate an amplification curve that crosses the threshold, then contamination occurred during the DNA extraction process, and replacement of extraction reagents with new ones and cleaning of workspace and pipettes is recommended. Positive control reactions should be performed using DNA from known positive isolates. Dilute the positive control DNA in order to decrease the likelihood of contamination of the PCR workspace, thereby avoiding false positives. In summary, for each target gene to be detected (i.e., for each mix that is made up), the following controls should be run:
- No-template negative controls (NTCs), at least in duplicate
- Extracted-water negative controls for DNA prep equipment and reagent cross-contamination, in duplicate
- Positive control, using prepared DNA from a known isolate and running RNAse P when using clinical specimens
A sample spreadsheet depicting the set-up of a 96-well real-time PCR plate is pictured below (Figure 6):
Figure 6. Example PCR template sheet for the N. meningitidis, H. influenzae, and S. pneumoniae species-specific real-time PCR assays. This example template should provide all the information needed for the laboratorian to set up the real-time PCR assay. The template should include: the assay(s) being run, date performed, PCR machine used, file name, cycle conditions, strain DNA to use as positive controls, and any notes to assist the laboratorian. On the layout, each of the wells to be used on the 96-well plate should be clearly labeled with either the specimen name or type of control. In this case, three assays are being run on same plate and each assay is color-coded: sodC (or ctrA) for N. meningitidis detection in pale red; hpd for H. influenzae detection in green, and lytA for S. pneumoniae detection in blue. The non-template controls (ntc) for both the clean and dirty rooms are done in duplicate for each assay type (more can be added if the user deems it necessary). One positive control well should be used for each assay type and is labeled sodC +, hpd +, and lytA +. The water extraction to control for contamination during DNA extraction is labeled Extracted H2O. The 17 unknown specimens are labeled unk-1 to unk-17. The panel to the right depicts the amount of reagent needed for 1 reaction and also the amount needed to make a master mix of reagents for each assay being run. In this example, enough Master Mix (Mmix), which is the commercial mix of DNA polymerase, dNTPs, and buffer to be used, H2O, forward and reverse primers, and probe should be made for 25 reactions. The number 25 was calculated from the number of wells needed for each assay plus an additional 2 wells to ensure that the user has enough master mix...view larger
Real-time PCR protocol
- Prior to beginning, plan the experiment by filling out and printing a PCR template worksheet (see Figure 6 for an example). Also, be sure sufficient quantities of working stocks of primers and probes to be used are available.
- Turn on the real-time PCR machine and make sure lamp is warming up.
- Remove DNA preps and positive control DNA from -20°C to the dirty room/hood to thaw.
- In the clean room/hood, gather reagents: commercial PCR master mix, primers, probes, and PCR grade water. If they are used infrequently enough and are therefore stored at -20°C, allow working stocks of primers and probes to thaw completely before use.
Vortex or flick each tube before using. Assemble one master mix per primer and probe set to be used. For each extraction to be tested, the master mix should contain:
12.5 µl of master mix
4.5 µl of sterile, PCR-grade water
2 µl of forward primer
2 µl of reverse primer
2 µl of probe
23 µl total before adding 2 µl DNAWhen calculating volumes of master mix reagents, add enough master mix reagents for 2 extra reactions to ensure there will be enough mix.
- Pipette 23 µl of this master mix into each appropriate well of a 96-well plate, according to the plate template worksheet. Add 2 µl of PCR-grade water to the clean NTC wells and then cap only that row. Ensure that the lid is flush with the plate to avoid evaporation from wells during PCR.
- Incorrectly capped or uncapped wells, if run in the machine, will lead to reaction failures and could contaminate the machine. If this happens, wipe down the interior of the machine with 70% isopropanol.
- If available, cover plate with one-time use adhesive film. Wipe down the clean workspace with 10% bleach (10:1 water: concentrated bleach), then 70% ethanol and turn on UV light for 1 hour, if available. Remove laboratory coat and gloves. Put on a fresh pair of gloves. Carefully transport the plate to the dirty room/hood.
- Place the plate in the dirty room/hood. Put on a new laboratory coat and keep the same pair of gloves on. If one was used, remove adhesive cover from plate.
- According to your template worksheet, add 2 µl to the appropriate well of the following in this order:
- Template DNA
- Extracted water controls
- Dirty NTCs
- Positive control DNA
- Cap columns of wells as you go, capping NTC control wells last. Use the roller tool to secure caps tightly.
- Wipe down the workspace with 10% bleach, then 70% ethanol and turn on the UV light for 1 hour, if available. Remove laboratory coat and gloves and discard the gloves.
- If possible, spin the plate at 500 x g for a few seconds to bring down any droplets and to mix. Transport the plate directly to and place it in the real-time PCR machine.
- Follow the instructions for machine operation that were provided by the manufacturer. Make sure that the machine is set to read the fluorescence of the reference dye contained in the commercial master mix, which is often ROX, in addition to the dye conjugated to each probe used (e.g., FAM, HEX, CY5). The cycle parameters suggested for the primers and probes given in the table are:
- 1 cycle of 50°C for 2 minutes
- 1 cycle of 95°C for 10 minutes
- 50 cycles of 95°C for 15 seconds + 60°C for 1 minute
- Turn off the machine lamp when the assay is complete.
- Data analysis
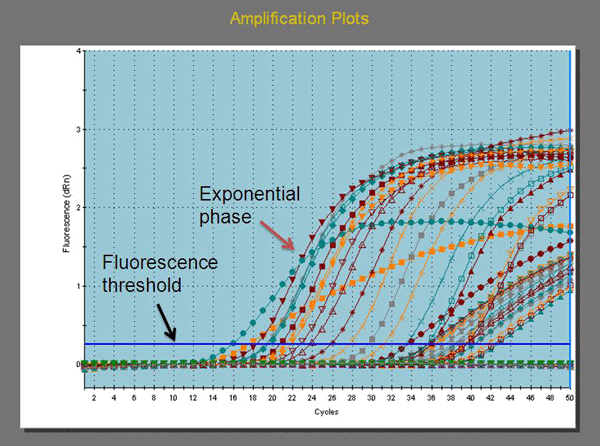
Figure 7. Amplification plot generated by a real-time PCR assay. This is a graph showing a plot of amplification cycle numbers on the X axis versus fluorescence units (dRn) on the Y axis for each reaction. dRn is the baseline subtracted fluorescent reading normalized to the reference dye. The green line of the fluorescence threshold is highlighted by the black arrow and the exponential phase is highlighted by the red arrow.
The readout of the data generated by real-time PCR machines will come in two main formats: amplification plots (Figure 7) and plate sample values. The curves that are generated should be sigmoidal in shape, ideally plateauing as the last cycle is approached, indicating complete use of reactants. The cycle number at which the fluorescence curve for each sample crosses the fluorescence threshold (green line in the above graph, which is generated automatically by the data analysis software) is referred to as the cycle threshold value, or Ct value. The plate sample values format of data readout is simply a listing of the Ct value generated by each reaction. The fluorescence threshold should be set higher than the negative controls and negative specimens and should be within the start of the exponential phase (see Figure 7). In an optimal PCR reaction running at 100% efficiency, exact doubling of the PCR product occurs at every cycle during the exponential phase. The laboratorian should carefully examine each curve and document all Ct values obtained. If possible, it is recommended that the electronic data files be saved for each run of the real-time PCR machine, including amplification plots for future reference.
As with any experiment, results for the unknowns cannot be evaluated without first assessing the results for the controls. NTCs and negative controls should give "No Ct" and should produce amplification curves that are straight lines near zero. Positive controls should give Ct values less than 35, and the amplifications plots of the positive controls and any unknown specimens that generate curves should all be sigmoidal. The commercial master mix used may contain a reference dye, which is often ROX. If the real-time PCR machine is working properly, the amplification plot and plate sample value for ROX in each reaction should resemble those of the NTCs and negative controls. If these criteria are not met, it will be difficult to interpret the results of the unknown samples. If this is the case, refer to the troubleshooting section below.
When determining whether to call an unknown specimen positive or negative for an etiology, the following cutoff values are used in the Meningitis Laboratory at CDC:
Positive = Ct ≤ 35
Negative = Ct > 40
Equivocal = Ct 36-40
- Ct values ≤ 35 are considered positive and Ct values > 40 are considered negative. Those with Ct values between 36 and 40 are considered equivocal and should be retested after diluting the template DNA 1:4 and 1:10 in PCR-grade water to reduce any inhibitors that may be interfering with the reaction. If the Ct value decreases to ≤35, the specimen should be considered positive.
- The amplification plots should be analyzed to ensure they are smooth and sigmoidal in shape. If a plot is lacking these characteristics, it should be considered negative or retested.
- Other troubleshooting suggestions are described below.
- Labs that are starting real-time PCR programs should try to pair with a reference lab to help with QA/QC.
Commonly Used Terms
- Reporter dye/fluorophore: Used to monitor PCR product accumulation.
- Quencher dye: Absorbs the light energy of the excited-state reporter dye.
- Reference dye: A dye, commonly ROX, which fluoresces at a constant level during the reaction. It is used to normalize the fluorescent signal of the reporter dye. Some systems do not use a reference dye.
- Ct or cycle threshold: The PCR cycle number at which fluorescence measured by the instrument is at a statistically significant level above background. The Ct is inversely proportional to the log of the initial copy number.
- Exponential phase: Phase at which exact doubling of the PCR product is accumulating at every cycle, assuming 100 % reaction efficiency. Exponential amplification occurs because all of the reagents are fresh and available and the kinetics of the reaction push the reaction to favor doubling of amplicon.
- R: Raw fluorescence reading in arbitrary units
- Rn: Fluorescent reading normalized to the reference dye
- dRn, ΔRn: Baseline subtracted fluorescent reading normalized to the reference dye
- NTC (non-template control): A sample type containing all the reaction components except the DNA template. Instead of template DNA, 2 µl of sterile, PCR-grade water is added to the NTC reaction wells. Water added in the clean room is called "clean NTC" and water added in the dirty room is called "dirty NTC".
- Amplification plot: Graph showing a plot of amplification cycle numbers on the X axis versus fluorescence units on the Y axis for each reaction
- Workstation for real-time PCR reaction set-up
- Workflow for detection of bacterial meningitis pathogens by PCR
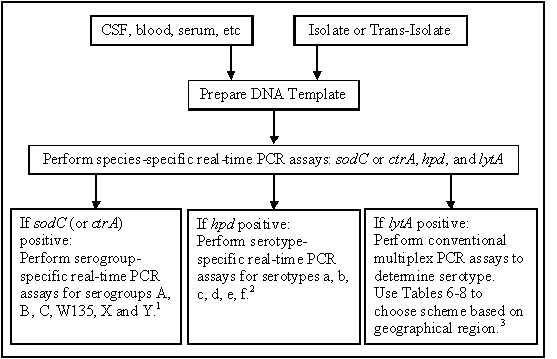
Figure 8. Workflow for detection and characterization of bacterial meningitis pathogens by PCR. Once DNA template is prepared from either a clinical specimen, an isolate, or from sampling a bottle of inoculated Tran-Isolate medium, the species-specific real-time PCR assays should be run. Any reactions positive for N. meningitis, H. influenzae, or S. pneumoniae should be further characterized using the appropriate serogrouping or serotyping PCR assay.
1 It is not always practical to test for all serogroups for which assays are available in a laboratory. Testing algorithms may be set up in laboratories with previous knowledge of the predominance or lack of serogroups within that particular geographic region to test for the most common serogroups first. Modifications may be made to the testing algorithm for any laboratory based on information about current strains that are circulating in the region. For example, in Africa, testing to detect serogroups A and W135 (and X in some regions) should be adequate to characterize most specimens. Specimens reacting negatively in the A and W135 assays should then be tested using the other available assays, particularly C, Y, X and B. Nearly all invasive specimens are serogroupable, if they are tested against a comprehensive panel of assays and proper controls are used.
2 It is not always practical to test for all serotypes for which assays are available in a laboratory. If a Hib vaccination program does not exist or is fairly new, testing all hpd positive specimens for Hib first may be adequate to characterize most specimens.
3 For bacterial isolates confirmed as S. pneumoniae by microbiological methods, conventional multiplex PCR serotyping is used. For clinical specimens that are lytA-positive by real-time PCR and have Ct values ≤30, conventional multiplex PCR serotyping can be used. If Ct values are >30, then a real-time PCR serotyping approach is recommended for increased sensitivity. - Troubleshooting
- Are separate rooms, laboratory coats, gloves, and pipettes being used for master mix preparation versus template addition?
- Are sterile barrier-filtered tips being used?
- Are bleach and ethanol being used to clean work areas?
- Reaction tubes should not be re-opened and should be immediately discarded.
- Reaction tubes should be protected during transport from the clean room to the dirty room.
- Consumable reagents (tips, plates, caps, adhesive films, etc.) should not be reused.
- The amount of positive control DNA should be limited.
If there are high or no Ct counts in known positives, consider:
- Is specimen handling questioned? Was there sufficient specimen volume?
- Clinical specimens should ideally be shipped on dry ice and frozen at -70°C.
- The sample volume should ideally be at least 200 µl for optimal yields.
- Was the DNA extraction process performed correctly?
- If mutanolysin and lysozyme are not used, DNA from gram-positive bacteria may not be efficiently extracted and thus will not be detectable.
- Could any of the extraction reagents, supplies, or workspaces be contaminated?
- Are poor DNA integrity and/or inhibitors suspected?
- Dilute the template DNA 1:4 and 1:10 and repeating the reaction. If Ct values decrease compared to undiluted, this can be indicative of an inhibitor in the DNA preparation. The specimen should be considered positive if the Ct count is less than 35 upon repeat.
- Buffers containing phosphate such as phosphate buffered saline should be avoided as phosphate is a strong inhibitor of PCR.
- If possible, consider sending the specimen or extraction to a reference laboratory.
- Perform human RNAaseP assay to determine if extensive DNA degradation had occurred.
- A dilution of the DNA preparation should also be assayed for RNAaseP to determine if inhibitors are present.
- Did a power failure or electrical current surge affect your results?
- It is strongly recommended that the real-time PCR machine and associated computer be plugged directly into a surge protector with a battery back-up.
If problems persist, contact an experienced reference laboratory for assistance.
Recommended for further reading: (7, 46).
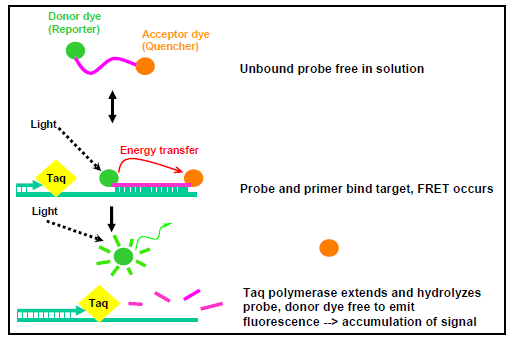{kind=link}
{kind=link}
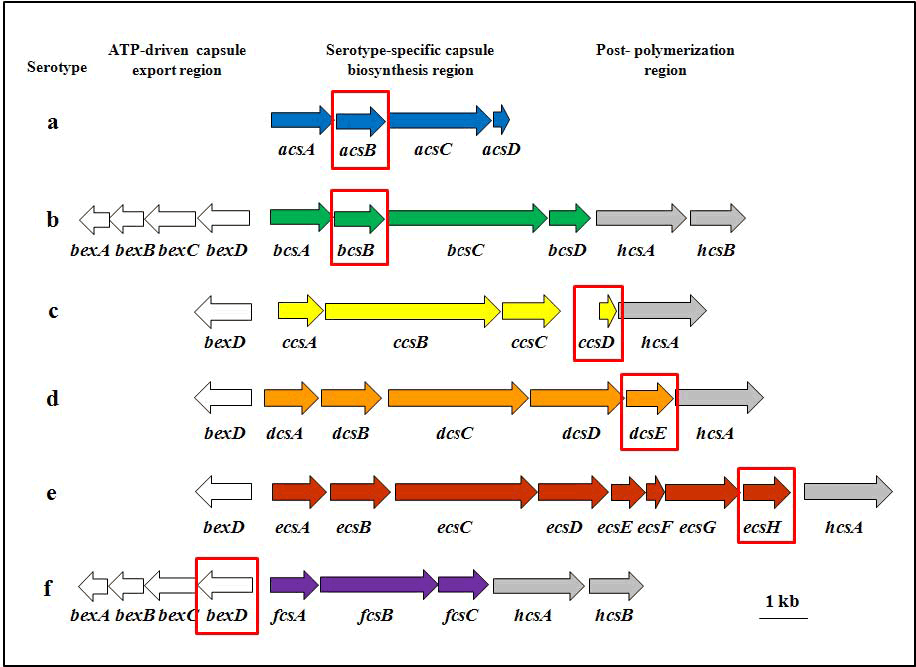{kind=link}
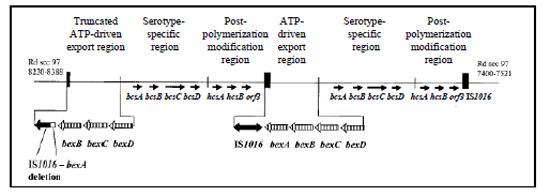{kind=link}
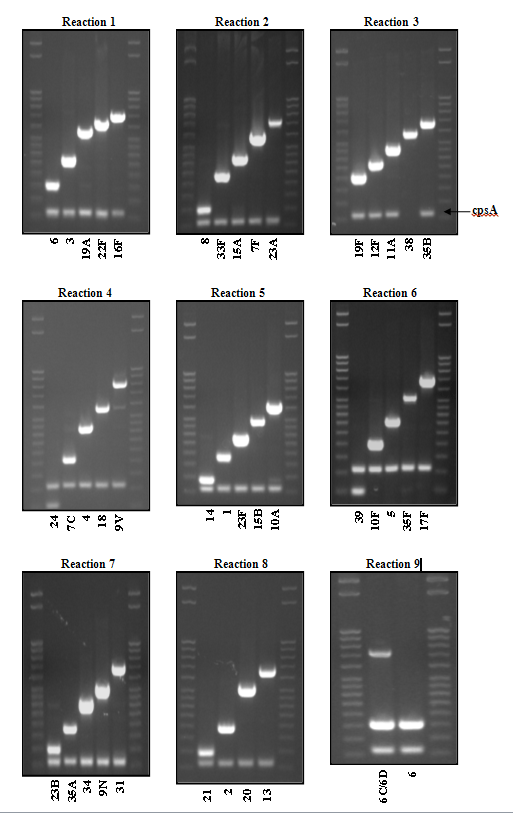{kind=link}
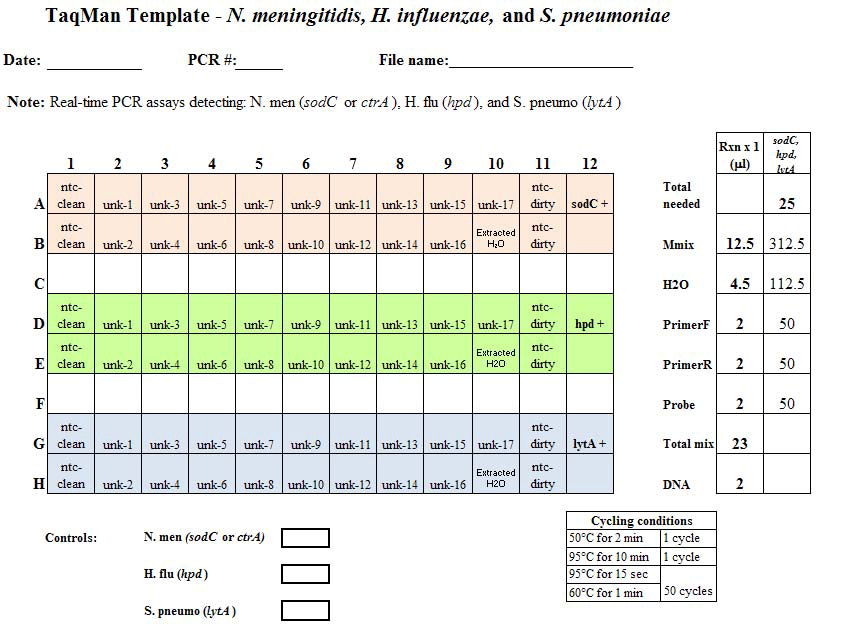{kind=link}
References
- Bentley, S. D., G. S. Vernikos, L. A. Snyder, C. Churcher, C. Arrowsmith, T. Chillingworth, A. Cronin, P. H. Davis, N. E. Holroyd, K. Jagels, M. Maddison, S. Moule, E. Rabbinowitsch, S. Sharp, L. Unwin, S. Whitehead, M. A. Quail, M. Achtman, B. Barrell, N. J. Saunders, and J. Parkhill. 2007. Meningococcal genetic variation mechanisms viewed through comparative analysis of serogroup C strain FAM18. PLoS Genetics 3:e23.
- Boisier, P., P. Nicolas, S. Djibo, M. K. Taha, I. Jeanne, H. B. Mainassara, B. Tenebray, K. K. Kairo, D. Giorgini, and S. Chanteau. 2007. Meningococcal meningitis: unprecedented incidence of serogroup X-related cases in 2006 in Niger. Clinical Infectious Diseases 44:657-663.
- Borel, T., A. M. Rose, M. Guillerm, F. Sidikou, S. Gerstl, A. Djibo, N. Nathan, S. Chanteau, and P. J. Guerin. 2006. High sensitivity and specificity of the Pastorex latex agglutination test for Neisseria meningitidis serogroup A during a clinical trial in Niger. Transactions of the Royal Society of Tropical Medicine and Hygiene 100:964-969.
- Borrow, R., H. Claus, U. Chaudhry, M. Guiver, E. B. Kaczmarski, M. Frosch, and A. J. Fox. 1998. siaD PCR ELISA for confirmation and identification of serogroup Y and W135 meningococcal infections. FEMS Microbiology Letters 159:209-214.
- Borrow, R., H. Claus, M. Guiver, L. Smart, D. M. Jones, E. B. Kaczmarski, M. Frosch, and A. J. Fox. 1997. Non-culture diagnosis and serogroup determination of meningococcal B and C infection by a sialyltransferase (siaD) PCR ELISA. Epidemiology and Infection 118:111-117.
- Bundle, D. R., H. J. Jennings, and C. P. Kenney. 1974. Studies on the group-specific polysaccharide of Neisseria meningitidis serogroup X and an improved procedure for its isolation. Journal of Biological Chemistry 249:4797-4801.
- Bustin, S. A. 2004. A-Z of quantitative PCR. International University Line, La Jolla, CA.
- Carvalho, M. G., M. L. Tondella, K. McCaustland, L. Weidlich, L. McGee, L. W. Mayer, A. Steigerwalt, M. Whaley, R. R. Facklam, B. Fields, G. Carlone, E. W. Ades, R. Dagan, and J. S. Sampson. 2007. Evaluation and improvement of real-time PCR assays targeting lytA, ply, and psaA genes for detection of pneumococcal DNA. Journal of Clinical Microbiology 45:2460-2466.
- Claus, H., R. Borrow, M. Achtman, G. Morelli, C. Kantelberg, E. Longworth, M. Frosch, and U. Vogel. 2004. Genetics of capsule O-acetylation in serogroup C, W-135, and Y meningococci. Molecular Microbiology 51:227-239.
- Claus, H., M. C. Maiden, R. Maag, M. Frosch, and U. Vogel. 2002. Many carried meningococci lack the genes required for capsule synthesis and transport. Microbiology 148:1813-1819.
- Claus, H., U. Vogel, M. Muhlenhoff, R. Gerardy-Schahn, and M. Frosch. 1997. Molecular divergence of the sia locus in different serogroups of Neisseria meningitidis expressing polysialic acid capsules. Molecular and General Genetics 257:28-34.
- Corless, C. E., M. Guiver, R. Borrow, V. Edwards-Jones, A. J. Fox, and E. B. Kaczmarski. 2001. Simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae in suspected cases of meningitis and septicemia using real-time PCR. Journal of Clinical Microbiology 39:1553-1558.
- Csako, G. 2006. Present and future of rapid and/or high-throughput methods for nucleic acid testing. Clinica Chimica Acta 363:6-31.
- Dolan-Livengood, J. M., Y. K. Miller, L. E. Martin, R. Urwin, and D. S. Stephens. 2003. Genetic basis for nongroupable Neisseria meningitidis. Journal of Infectious Diseases 187:1616-1628.
- Dolan, J. M., X. Wang, J. Theodore, C. Hatcher, M. G. Carvalho, B. Harcourt, F. C. Pimenta, B. Beall, K. Edmond, J. Mendisaihan, D. Altantsetseg, P. Nymadawa, M. Bakir, D. Toprak, A. Soysal, N. Messonnier, and L. W. Mayer. 2008. Presented at the 16th International Pathogenic Neisseria Conference, Rotterdam, The Netherlands.
- Edwards, U., A. Muller, S. Hammerschmidt, R. Gerardy-Schahn, and M. Frosch. 1994. Molecular analysis of the biosynthesis pathway of the alpha-2,8 polysialic acid capsule by Neisseria meningitidis serogroup B. Molecular Microbiology 14:141-149.
- Falla, T. J., D. W. Crook, L. N. Brophy, D. Maskell, J. S. Kroll, and E. R. Moxon. 1994. PCR for capsular typing of Haemophilus influenzae. Journal of Clinical Microbiology 32:2382-2386.
- Frosch, M., U. Edwards, K. Bousset, B. Krausse, and C. Weisgerber. 1991. Evidence for a common molecular origin of the capsule gene loci in gram-negative bacteria expressing group II capsular polysaccharides. Molecular Microbiology 5:1251-1263.
- Frosch, M., and A. Muller. 1993. Phospholipid substitution of capsular polysaccharides and mechanisms of capsule formation in Neisseria meningitidis. Molecular Microbiology 8:483-493.
- Gagneux, S. P., A. Hodgson, T. A. Smith, T. Wirth, I. Ehrhard, G. Morelli, B. Genton, F. N. Binka, M. Achtman, and G. Pluschke. 2002. Prospective study of a serogroup X Neisseria meningitidis outbreak in northern Ghana. Journal of Infectious Diseases 185:618-626.
- Ganguli, S., G. Zapata, T. Wallis, C. Reid, G. Boulnois, W. F. Vann, and I. S. Roberts. 1994. Molecular cloning and analysis of genes for sialic acid synthesis in Neisseria meningitidis group B and purification of the meningococcal CMP-NeuNAc synthetase enzyme. Journal of Bacteriology 176:4583-4589.
- Hammerschmidt, S., C. Birkholz, U. Zahringer, B. D. Robertson, J. van Putten, O. Ebeling, and M. Frosch. 1994. Contribution of genes from the capsule gene complex (cps) to lipooligosaccharide biosynthesis and serum resistance in Neisseria meningitidis. Molecular Microbiology 11:885-896.
- Hammerschmidt, S., A. Muller, H. Sillmann, M. Muhlenhoff, R. Borrow, A. Fox, J. van Putten, W. D. Zollinger, R. Gerardy-Schahn, and M. Frosch. 1996. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): correlation with bacterial invasion and the outbreak of meningococcal disease. Molecular Microbiology 20:1211-1220.
- Janson, H., M. Ruan, and A. Forsgren. 1993. Limited diversity of the protein D gene (hpd) among encapsulated and nonencapsulated Haemophilus influenzae strains. Infection and Immunity 61:4546-4552.
- Kroll, J. S. 1992. The genetics of encapsulation in Haemophilus influenzae. Journal of Infectious Diseases 165 Suppl 1:S93-96.
- Kroll, J. S., B. M. Loynds, and E. R. Moxon. 1991. The Haemophilus influenzae capsulation gene cluster: a compound transposon. Molecular Microbiology 5:1549-1560.
- Kroll, J. S., and E. R. Moxon. 1988. Capsulation and gene copy number at the cap locus of Haemophilus influenzae type b. Journal of Bacteriology 170:859-864.
- Kroll, J. S., S. Zamze, B. Loynds, and E. R. Moxon. 1989. Common organization of chromosomal loci for production of different capsular polysaccharides in Haemophilus influenzae. Journal of Bacteriology 171:3343-3347.
- LaClaire, L. L., M. L. Tondella, D. S. Beall, C. A. Noble, P. L. Raghunathan, N. E. Rosenstein, and T. Popovic. 2003. Identification of Haemophilus influenzae serotypes by standard slide agglutination serotyping and PCR-based capsule typing. Journal of Clinical Microbiology 41:393-396.
- Lee, L. G., C. R. Connell, and W. Bloch. 1993. Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Research 21:3761-3766.
- Liu, T. Y., E. C. Gotschlich, E. K. Jonssen, and J. R. Wysocki. 1971. Studies on the meningococcal polysaccharides. I. Composition and chemical properties of the group A polysaccharide. Journal of Biological Chemistry 246:2849-2858.
- Masson, L., and B. E. Holbein. 1983. Physiology of sialic acid capsular polysaccharide synthesis in serogroup B Neisseria meningitidis. Journal of Bacteriology 154:728-736.
- Messmer, T. O., J. S. Sampson, A. Stinson, B. Wong, G. M. Carlone, and R. R. Facklam. 2004. Comparison of four polymerase chain reaction assays for specificity in the identification of Streptococcus pneumoniae. Diagnostic Microbiology and Infectious Disease 49:249-254.
- Moore, C. E., A. Sengduangphachanh, T. Thaojaikong, J. Sirisouk, D. Foster, R. Phetsouvanh, L. McGee, D. W. Crook, P. N. Newton, and S. J. Peacock. 2010. Enhanced determination of Streptococcus pneumoniae serotypes associated with invasive disease in Laos by using a real-time polymerase chain reaction serotyping assay with cerebrospinal fluid. American Journal of Tropical Medicine and Hygiene 83:451-457.
- Mothershed, E. A., C. T. Sacchi, A. M. Whitney, G. A. Barnett, G. W. Ajello, S. Schmink, L. W. Mayer, M. Phelan, T. H. Taylor, Jr., S. A. Bernhardt, N. E. Rosenstein, and T. Popovic. 2004. Use of real-time PCR to resolve slide agglutination discrepancies in serogroup identification of Neisseria meningitidis. Journal of Clinical Microbiology 42:320-328.
- Mullis, K. B., and F. A. Faloona. 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods In Enzymology 155:335-350.
- Murdoch, D. R., T. P. Anderson, K. A. Beynon, A. Chua, A. M. Fleming, R. T. Laing, G. I. Town, G. D. Mills, S. T. Chambers, and L. C. Jennings. 2003. Evaluation of a PCR assay for detection of Streptococcus pneumoniae in respiratory and nonrespiratory samples from adults with community-acquired pneumonia. Journal of Clinical Microbiology 41:63-66.
- Pai, R., R. E. Gertz, and B. Beall. 2006. Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. Journal of Clinical Microbiology 44:124-131.
- Parkhill, J., M. Achtman, K. D. James, S. D. Bentley, C. Churcher, S. R. Klee, G. Morelli, D. Basham, D. Brown, T. Chillingworth, R. M. Davies, P. Davis, K. Devlin, T. Feltwell, N. Hamlin, S. Holroyd, K. Jagels, S. Leather, S. Moule, K. Mungall, M. A. Quail, M. A. Rajandream, K. M. Rutherford, M. Simmonds, J. Skelton, S. Whitehead, B. G. Spratt, and B. G. Barrell. 2000. Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature 404:502-506.
- Resti, M., M. Moriondo, M. Cortimiglia, G. Indolfi, C. Canessa, L. Becciolini, E. Bartolini, F. M. de Benedictis, M. de Martino, and C. Azzari. 2010. Community-acquired bacteremic pneumococcal pneumonia in children: diagnosis and serotyping by real-time polymerase chain reaction using blood samples. Clinical Infectious Diseases 51:1042-1049.
- Sadler, F., A. Fox, K. Neal, M. Dawson, K. Cartwright, and R. Borrow. 2003. Genetic analysis of capsular status of meningococcal carrier isolates. Epidemiology and Infection 130:59-70.
- Saiki, R. K., S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich, and N. Arnheim. 1985. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230:1350-1354.
- Satola, S. W., P. L. Schirmer, and M. M. Farley. 2003. Complete sequence of the cap locus of Haemophilus influenzae serotype b and nonencapsulated b capsule-negative variants. Infection and Immunity 71:3639-3644.
- Satola, S. W., P. L. Schirmer, and M. M. Farley. 2003. Genetic analysis of the capsule locus of Haemophilus influenzae serotype f. Infection and Immunity 71:7202-7207.
- Song, X. M., A. Forsgren, and H. Janson. 1995. The gene encoding protein D (hpd) is highly conserved among Haemophilus influenzae type b and nontypeable strains. Infection and Immunity 63:696-699.
- Stratagene. 2006. Introduction to Quantitative PCR: Methods and Application Guide.
- Suzuki, N., M. Yuyama, S. Maeda, H. Ogawa, K. Mashiko, and Y. Kiyoura. 2006. Genotypic identification of presumptive Streptococcus pneumoniae by PCR using four genes highly specific for S. pneumoniae. Journal of Medical Microbiology 55:709-714.
- Swartley, J. S., J. H. Ahn, L. J. Liu, C. M. Kahler, and D. S. Stephens. 1996. Expression of sialic acid and polysialic acid in serogroup B Neisseria meningitidis: divergent transcription of biosynthesis and transport operons through a common promoter region. Journal of Bacteriology 178:4052-4059.
- Swartley, J. S., L. J. Liu, Y. K. Miller, L. E. Martin, S. Edupuganti, and D. S. Stephens. 1998. Characterization of the gene cassette required for biosynthesis of the (alpha1-->6)-linked N-acetyl-D-mannosamine-1-phosphate capsule of serogroup A Neisseria meningitidis. Journal of Bacteriology 180:1533-1539.
- Swartley, J. S., A. A. Marfin, S. Edupuganti, L. J. Liu, P. Cieslak, B. A. Perkins, J. D. Wenger, and D. S. Stephens. 1997. Capsule switching of Neisseria meningitidis. Proceedings of the National Academy of Sciences of the United States of America 94:271-276.
- Swartley, J. S., and D. S. Stephens. 1994. Identification of a genetic locus involved in the biosynthesis of N-acetyl-D-mannosamine, a precursor of the (alpha 2-->8)-linked polysialic acid capsule of serogroup B Neisseria meningitidis. Journal of Bacteriology 176:1530-1534.
- Taha, M.-K. 2000. Simultaneous approach for nonculture PCR-based identification and serogroup prediction of Neisseria meningitidis. Journal of Clinical Microbiology 38:855-857.
- Taha, M. K., J. M. Alonso, M. Cafferkey, D. A. Caugant, S. C. Clarke, M. A. Diggle, A. Fox, M. Frosch, S. J. Gray, M. Guiver, S. Heuberger, J. Kalmusova, K. Kesanopoulos, A. M. Klem, P. Kriz, J. Marsh, P. Mölling, K. Murphy, P. Olcén, O. Sanou, G. Tzanakaki, and U. Vogel. 2005. Interlaboratory comparison of PCR-based identification and genogrouping of Neisseria meningitidis. Journal of Clinical Microbiology 43:144-149.
- Tarrago, D., A. Fenoll, D. Sanchez-Tatay, L. A. Arroyo, C. Munoz-Almagro, C. Esteva, W. P. Hausdorff, J. Casal, and I. Obando. 2008. Identification of pneumococcal serotypes from culture-negative clinical specimens by novel real-time PCR. Clinical Microbiology and Infection 14:828-834.
- Tettelin, H., N. J. Saunders, J. Heidelberg, A. C. Jeffries, K. E. Nelson, J. A. Eisen, K. A. Ketchum, D. W. Hood, J. F. Peden, R. J. Dodson, W. C. Nelson, M. L. Gwinn, R. DeBoy, J. D. Peterson, E. K. Hickey, D. H. Haft, S. L. Salzberg, O. White, R. D. Fleischmann, B. A. Dougherty, T. Mason, A. Ciecko, D. S. Parksey, E. Blair, H. Cittone, E. B. Clark, M. D. Cotton, T. R. Utterback, H. Khouri, H. Qin, J. Vamathevan, J. Gill, V. Scarlato, V. Masignani, M. Pizza, G. Grandi, L. Sun, H. O. Smith, C. M. Fraser, E. R. Moxon, R. Rappuoli, and J. C. Venter. 2000. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 287:1809-1815.
- Tzeng, Y.-L., C. Noble, and D. S. Stephens. 2003. Genetic basis for biosynthesis of the (alpha 1-->4)-linked N-acetyl-D-glucosamine 1-phosphate capsule of Neisseria meningitidis serogroup X. Infection and Immunity 71:6712-6720.
- Tzeng, Y. L., A. K. Datta, C. A. Strole, M. A. Lobritz, R. W. Carlson, and D. S. Stephens. 2005. Translocation and surface expression of lipidated serogroup B capsular Polysaccharide in Neisseria meningitidis. Infection and Immunity 73:1491-1505.
- Vogel, U., H. Claus, and M. Frosch. 2000. Rapid serogroup switching in Neisseria meningitidis. New England Journal of Medicine 342:219-220.
- Waggoner-Fountain, L. A., J. O. Hendley, E. J. Cody, V. A. Perriello, and L. G. Donowitz. 1995. The emergence of Haemophilus influenzae types e and f as significant pathogens. Clinical Infectious Diseases 21:1322-1324.
- Wang, X., R. Mair, C. Hatcher, M. J. Theodore, K. Edmond, H. M. Wu, B. H. Harcourt, G. Carvalho Mda, F. Pimenta, P. Nymadawa, D. Altantsetseg, M. Kirsch, S. W. Satola, A. Cohn, N. E. Messonnier, and L. W. Mayer. 2011. Detection of bacterial pathogens in Mongolia meningitis surveillance with a new real-time PCR assay to detect Haemophilus influenzae. International Journal of Medical Microbiology 301:303-309.
- Weber, M. V., H. Claus, M. C. Maiden, M. Frosch, and U. Vogel. 2006. Genetic mechanisms for loss of encapsulation in polysialyltransferase-gene-positive meningococci isolated from healthy carriers. International Journal of Medical Microbiology 296:475-484.
- Whatmore, A. M., A. Efstratiou, A. P. Pickerill, K. Broughton, G. Woodard, D. Sturgeon, R. George, and C. G. Dowson. 2000. Genetic relationships between clinical isolates of Streptococcus pneumoniae, Streptococcus oralis, and Streptococcus mitis: Characterization of “atypical” pneumococci and organisms allied to S. mitis harboring S. pneumoniae virulence factor-encoding genes. Infection and Immunity 68:1374–1382.
- Page last reviewed: April 15, 2016
- Page last updated: March 15, 2012
- Content source: