Neoplasm
A neoplasm (/ˈniːoʊplæzəm, ˈniə-/[1]) is a type of abnormal and excessive growth, called neoplasia, of tissue. The growth of a neoplasm is uncoordinated with that of the normal surrounding tissue, and it persists growing abnormally, even if the original trigger is removed.[2][3][4] This abnormal growth usually (but not always) forms a mass.[5] When it forms a mass, it may be called a tumor.
| Neoplasm | |
|---|---|
| Other names | Tumor, tumour, carcinocytes |
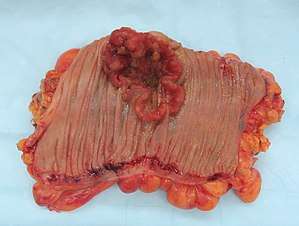 | |
| Colectomy specimen containing a malignant neoplasm, namely an invasive example of colorectal cancer (the crater-like, reddish, irregularly shaped tumor) | |
| Specialty | Oncology |
ICD-10 classifies neoplasms into four main groups: benign neoplasms, in situ neoplasms, malignant neoplasms, and neoplasms of uncertain or unknown behavior.[6] Malignant neoplasms are also simply known as cancers and are the focus of oncology.
Prior to the abnormal growth of tissue, as neoplasia, cells often undergo an abnormal pattern of growth, such as metaplasia or dysplasia.[7] However, metaplasia or dysplasia does not always progress to neoplasia.[2] The word is from Ancient Greek νέος- neo ("new") and πλάσμα plasma ("formation", "creation").
Types
| -plasia and -trophy |
|---|
|
|
A neoplasm can be benign, potentially malignant, or malignant (cancer).[8]
- Benign tumors include uterine fibroids, osteophytes and melanocytic nevi (skin moles). They are circumscribed and localized and do not transform into cancer.[7]
- Potentially-malignant neoplasms include carcinoma in situ. They are localised, do not invade and destroy but in time, may transform into a cancer.
- Malignant neoplasms are commonly called cancer. They invade and destroy the surrounding tissue, may form metastases and, if untreated or unresponsive to treatment, will generally prove fatal.
- Secondary neoplasm refers to any of a class of cancerous tumor that is either a metastatic offshoot of a primary tumor, or an apparently unrelated tumor that increases in frequency following certain cancer treatments such as chemotherapy or radiotherapy.
- Rarely there can be a metastatic neoplasm with no known site of the primary cancer and this is classed as a cancer of unknown primary origin
Clonality
Neoplastic tumors are often heterogeneous and contain more than one type of cell, but their initiation and continued growth is usually dependent on a single population of neoplastic cells. These cells are presumed to be clonal – that is, they are derived from the same cell,[9] and all carry the same genetic or epigenetic anomaly – evident of clonality. For lymphoid neoplasms, e.g. lymphoma and leukemia, clonality is proven by the amplification of a single rearrangement of their immunoglobulin gene (for B cell lesions) or T cell receptor gene (for T cell lesions). The demonstration of clonality is now considered to be necessary to identify a lymphoid cell proliferation as neoplastic.[10]
It is tempting to define neoplasms as clonal cellular proliferations but the demonstration of clonality is not always possible. Therefore, clonality is not required in the definition of neoplasia.
Neoplasm vs. tumor
The word tumor or tumour comes from the Latin word for swelling, which is one of the cardinal signs of inflammation. The word originally referred to any form of swelling, neoplastic or not. In modern English, tumor is used as a synonym for neoplasm (a solid or fluid-filled cystic lesion that may or may not be formed by an abnormal growth of neoplastic cells) that appears enlarged in size.[11][12] Some neoplasms do not form a tumor - these include leukemia and most forms of carcinoma in situ. Tumor is also not synonymous with cancer. While cancer is by definition malignant, a tumor can be benign, precancerous, or malignant.
The terms mass and nodule are often used synonymously with tumor. Generally speaking, however, the term tumor is used generically, without reference to the physical size of the lesion.[2] More specifically, the term mass is often used when the lesion has a maximal diameter of at least 20 millimeters (mm) in greatest direction, while the term nodule is usually used when the size of the lesion is less than 20 mm in its greatest dimension (25.4 mm = 1 inch).[2]
Causes
A neoplasm can be caused by an abnormal proliferation of tissues, which can be caused by genetic mutations. Not all types of neoplasms cause a tumorous overgrowth of tissue, however (such as leukemia or carcinoma in situ) and similarities between neoplasmic growths and regenerative processes, e.g., dedifferentiation and rapid cell proliferation, have been pointed out.[13]
Recently, tumor growth has been studied using mathematics and continuum mechanics. Vascular tumors such as hemangiomas, and lymphangiomas, (formed from blood or lymph vessels) are thus looked at as being amalgams of a solid skeleton formed by sticky cells and an organic liquid filling the spaces in which cells can grow.[14] Under this type of model, mechanical stresses and strains can be dealt with and their influence on the growth of the tumor and the surrounding tissue and vasculature elucidated. Recent findings from experiments that use this model show that active growth of the tumor is restricted to the outer edges of the tumor, and that stiffening of the underlying normal tissue inhibits tumor growth as well.[15]
Benign conditions that are not associated with an abnormal proliferation of tissue (such as sebaceous cysts) can also present as tumors, however, but have no malignant potential. Breast cysts (as occur commonly during pregnancy and at other times) are another example, as are other encapsulated glandular swellings (thyroid, adrenal gland, pancreas).
Encapsulated hematomas, encapsulated necrotic tissue (from an insect bite, foreign body, or other noxious mechanism), keloids (discrete overgrowths of scar tissue) and granulomas may also present as tumors.
Discrete localized enlargements of normal structures (ureters, blood vessels, intrahepatic or extrahepatic biliary ducts, pulmonary inclusions, or gastrointestinal duplications) due to outflow obstructions or narrowings, or abnormal connections, may also present as a tumor. Examples are arteriovenous fistulae or aneurysms (with or without thrombosis), biliary fistulae or aneurysms, sclerosing cholangitis, cysticercosis or hydatid cysts, intestinal duplications, and pulmonary inclusions as seen with cystic fibrosis. It can be dangerous to biopsy a number of types of tumor in which the leakage of their contents would potentially be catastrophic. When such types of tumors are encountered, diagnostic modalities such as ultrasound, CT scans, MRI, angiograms, and nuclear medicine scans are employed prior to (or during) biopsy or surgical exploration/excision in an attempt to avoid such severe complications.
The nature of a tumor is determined by imaging, by surgical exploration, or by a pathologist after examination of the tissue from a biopsy or a surgical specimen.
Malignant neoplasms
DNA damage
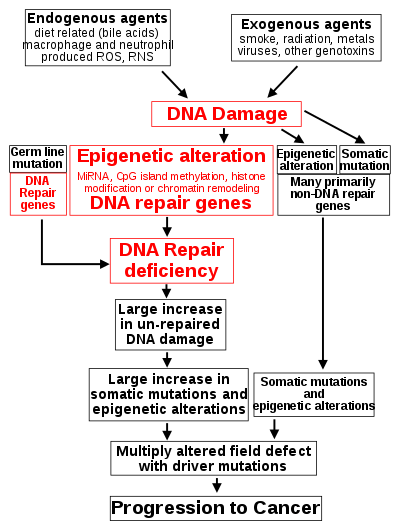
DNA damage is considered to be the primary underlying cause of malignant neoplasms known as cancers.[16] Its central role in progression to cancer is illustrated in the figure in this section, in the box near the top. (The central features of DNA damage, epigenetic alterations and deficient DNA repair in progression to cancer are shown in red.) DNA damage is very common. Naturally occurring DNA damages (mostly due to cellular metabolism and the properties of DNA in water at body temperatures) occur at a rate of more than 60,000 new damages, on average, per human cell, per day [also see article DNA damage (naturally occurring) ]. Additional DNA damages can arise from exposure to exogenous agents. Tobacco smoke causes increased exogenous DNA damage, and these DNA damages are the likely cause of lung cancer due to smoking.[17] UV light from solar radiation causes DNA damage that is important in melanoma.[18] Helicobacter pylori infection produces high levels of reactive oxygen species that damage DNA and contributes to gastric cancer.[19] Bile acids, at high levels in the colons of humans eating a high fat diet, also cause DNA damage and contribute to colon cancer.[20] Katsurano et al. indicated that macrophages and neutrophils in an inflamed colonic epithelium are the source of reactive oxygen species causing the DNA damages that initiate colonic tumorigenesis.[21] Some sources of DNA damage are indicated in the boxes at the top of the figure in this section.
Individuals with a germ line mutation causing deficiency in any of 34 DNA repair genes (see article DNA repair-deficiency disorder) are at increased risk of cancer. Some germ line mutations in DNA repair genes cause up to 100% lifetime chance of cancer (e.g., p53 mutations).[22] These germ line mutations are indicated in a box at the left of the figure with an arrow indicating their contribution to DNA repair deficiency.
About 70% of malignant neoplasms have no hereditary component and are called "sporadic cancers".[23] Only a minority of sporadic cancers have a deficiency in DNA repair due to mutation in a DNA repair gene. However, a majority of sporadic cancers have deficiency in DNA repair due to epigenetic alterations that reduce or silence DNA repair gene expression. For example, of 113 sequential colorectal cancers, only four had a missense mutation in the DNA repair gene MGMT, while the majority had reduced MGMT expression due to methylation of the MGMT promoter region (an epigenetic alteration).[24] Five reports present evidence that between 40% and 90% of colorectal cancers have reduced MGMT expression due to methylation of the MGMT promoter region.[25][26][27][28][29]
Similarly, out of 119 cases of mismatch repair-deficient colorectal cancers that lacked DNA repair gene PMS2 expression, PMS2 was deficient in 6 due to mutations in the PMS2 gene, while in 103 cases PMS2 expression was deficient because its pairing partner MLH1 was repressed due to promoter methylation (PMS2 protein is unstable in the absence of MLH1).[30] In the other 10 cases, loss of PMS2 expression was likely due to epigenetic overexpression of the microRNA, miR-155, which down-regulates MLH1.[31]
In further examples, epigenetic defects were found at frequencies of between 13%-100% for the DNA repair genes BRCA1, WRN, FANCB, FANCF, MGMT, MLH1, MSH2, MSH4, ERCC1, XPF, NEIL1 and ATM. These epigenetic defects occurred in various cancers (e.g. breast, ovarian, colorectal and head and neck). Two or three deficiencies in expression of ERCC1, XPF or PMS2 occur simultaneously in the majority of the 49 colon cancers evaluated by Facista et al.[32] Epigenetic alterations causing reduced expression of DNA repair genes is shown in a central box at the third level from the top of the figure in this section, and the consequent DNA repair deficiency is shown at the fourth level.
When expression of DNA repair genes is reduced, DNA damages accumulate in cells at a higher than normal level, and these excess damages cause increased frequencies of mutation or epimutation. Mutation rates strongly increase in cells defective in DNA mismatch repair[33][34] or in homologous recombinational repair (HRR).[35]
During repair of DNA double strand breaks, or repair of other DNA damages, incompletely cleared sites of repair can cause epigenetic gene silencing.[36][37] DNA repair deficiencies (level 4 in the figure) cause increased DNA damages (level 5 in the figure) which result in increased somatic mutations and epigenetic alterations (level 6 in the figure).
Field defects, normal appearing tissue with multiple alterations (and discussed in the section below), are common precursors to development of the disordered and improperly proliferating clone of tissue in a malignant neoplasm. Such field defects (second level from bottom of figure) may have multiple mutations and epigenetic alterations.
Once a cancer is formed, it usually has genome instability. This instability is likely due to reduced DNA repair or excessive DNA damage. Because of such instability, the cancer continues to evolve and to produce sub clones. For example, a renal cancer, sampled in 9 areas, had 40 ubiquitous mutations, demonstrating tumor heterogeneity (i.e. present in all areas of the cancer), 59 mutations shared by some (but not all areas), and 29 “private” mutations only present in one of the areas of the cancer.[38]
Field defects
Various other terms have been used to describe this phenomenon, including "field effect", "field cancerization", and "field carcinogenesis". The term "field cancerization" was first used in 1953 to describe an area or "field" of epithelium that has been preconditioned by (at that time) largely unknown processes so as to predispose it towards development of cancer.[39] Since then, the terms "field cancerization" and "field defect" have been used to describe pre-malignant tissue in which new cancers are likely to arise.
Field defects are important in progression to cancer.[40][41] However, in most cancer research, as pointed out by Rubin[42] “The vast majority of studies in cancer research has been done on well-defined tumors in vivo, or on discrete neoplastic foci in vitro. Yet there is evidence that more than 80% of the somatic mutations found in mutator phenotype human colorectal tumors occur before the onset of terminal clonal expansion.[43] Similarly, Vogelstein et al.[44] point out that more than half of somatic mutations identified in tumors occurred in a pre-neoplastic phase (in a field defect), during growth of apparently normal cells. Likewise, epigenetic alterations present in tumors may have occurred in pre-neoplastic field defects.
An expanded view of field effect has been termed "etiologic field effect", which encompasses not only molecular and pathologic changes in pre-neoplastic cells but also influences of exogenous environmental factors and molecular changes in the local microenvironment on neoplastic evolution from tumor initiation to patient death.[45]
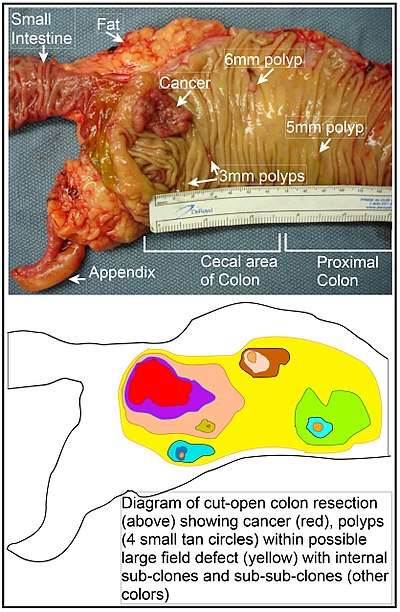
In the colon, a field defect probably arises by natural selection of a mutant or epigenetically altered cell among the stem cells at the base of one of the intestinal crypts on the inside surface of the colon. A mutant or epigenetically altered stem cell may replace the other nearby stem cells by natural selection. Thus, a patch of abnormal tissue may arise. The figure in this section includes a photo of a freshly resected and lengthwise-opened segment of the colon showing a colon cancer and four polyps. Below the photo there is a schematic diagram of how a large patch of mutant or epigenetically altered cells may have formed, shown by the large area in yellow in the diagram. Within this first large patch in the diagram (a large clone of cells), a second such mutation or epigenetic alteration may occur so that a given stem cell acquires an advantage compared to other stem cells within the patch, and this altered stem cell may expand clonally forming a secondary patch, or sub-clone, within the original patch. This is indicated in the diagram by four smaller patches of different colors within the large yellow original area. Within these new patches (sub-clones), the process may be repeated multiple times, indicated by the still smaller patches within the four secondary patches (with still different colors in the diagram) which clonally expand, until stem cells arise that generate either small polyps or else a malignant neoplasm (cancer).
In the photo, an apparent field defect in this segment of a colon has generated four polyps (labeled with the size of the polyps, 6mm, 5mm, and two of 3mm, and a cancer about 3 cm across in its longest dimension). These neoplasms are also indicated, in the diagram below the photo, by 4 small tan circles (polyps) and a larger red area (cancer). The cancer in the photo occurred in the cecal area of the colon, where the colon joins the small intestine (labeled) and where the appendix occurs (labeled). The fat in the photo is external to the outer wall of the colon. In the segment of colon shown here, the colon was cut open lengthwise to expose the inner surface of the colon and to display the cancer and polyps occurring within the inner epithelial lining of the colon.
If the general process by which sporadic colon cancers arise is the formation of a pre-neoplastic clone that spreads by natural selection, followed by formation of internal sub-clones within the initial clone, and sub-sub-clones inside those, then colon cancers generally should be associated with, and be preceded by, fields of increasing abnormality reflecting the succession of premalignant events. The most extensive region of abnormality (the outermost yellow irregular area in the diagram) would reflect the earliest event in formation of a malignant neoplasm.
In experimental evaluation of specific DNA repair deficiencies in cancers, many specific DNA repair deficiencies were also shown to occur in the field defects surrounding those cancers. The Table, below, gives examples for which the DNA repair deficiency in a cancer was shown to be caused by an epigenetic alteration, and the somewhat lower frequencies with which the same epigenetically caused DNA repair deficiency was found in the surrounding field defect.
| Cancer | Gene | Frequency in Cancer | Frequency in Field Defect | Ref. |
|---|---|---|---|---|
| Colorectal | MGMT | 46% | 34% | [25] |
| Colorectal | MGMT | 47% | 11% | [27] |
| Colorectal | MGMT | 70% | 60% | [46] |
| Colorectal | MSH2 | 13% | 5% | [27] |
| Colorectal | ERCC1 | 100% | 40% | [32] |
| Colorectal | PMS2 | 88% | 50% | [32] |
| Colorectal | XPF | 55% | 40% | [32] |
| Head and Neck | MGMT | 54% | 38% | [47] |
| Head and Neck | MLH1 | 33% | 25% | [48] |
| Head and Neck | MLH1 | 31% | 20% | [49] |
| Stomach | MGMT | 88% | 78% | [50] |
| Stomach | MLH1 | 73% | 20% | [51] |
| Esophagus | MLH1 | 77%-100% | 23%-79% | [52] |
Some of the small polyps in the field defect shown in the photo of the opened colon segment may be relatively benign neoplasms. Of polyps less than 10mm in size, found during colonoscopy and followed with repeat colonoscopies for 3 years, 25% were unchanged in size, 35% regressed or shrank in size while 40% grew in size.[53]
Genome instability
Cancers are known to exhibit genome instability or a mutator phenotype.[54] The protein-coding DNA within the nucleus is about 1.5% of the total genomic DNA.[55] Within this protein-coding DNA (called the exome), an average cancer of the breast or colon can have about 60 to 70 protein altering mutations, of which about 3 or 4 may be “driver” mutations, and the remaining ones may be “passenger” mutations[44] However, the average number of DNA sequence mutations in the entire genome (including non-protein-coding regions) within a breast cancer tissue sample is about 20,000.[56] In an average melanoma tissue sample (where melanomas have a higher exome mutation frequency[44]) the total number of DNA sequence mutations is about 80,000.[57] This compares to the very low mutation frequency of about 70 new mutations in the entire genome between generations (parent to child) in humans.[58][59]
The high frequencies of mutations in the total nucleotide sequences within cancers suggest that often an early alteration in the field defects giving rise to a cancer (e.g. yellow area in the diagram in this section) is a deficiency in DNA repair. The large field defects surrounding colon cancers (extending to at about 10 cm on each side of a cancer) were shown by Facista et al.[32] to frequently have epigenetic defects in 2 or 3 DNA repair proteins (ERCC1, XPF or PMS2) in the entire area of the field defect. Deficiencies in DNA repair cause increased mutation rates.[33][34][35] A deficiency in DNA repair, itself, can allow DNA damages to accumulate, and error-prone translesion synthesis past some of those damages may give rise to mutations. In addition, faulty repair of these accumulated DNA damages may give rise to epimutations. These new mutations or epimutations may provide a proliferative advantage, generating a field defect. Although the mutations/epimutations in DNA repair genes do not, themselves, confer a selective advantage, they may be carried along as passengers in cells when the cells acquire additional mutations/epimutations that do provide a proliferative advantage.
Etymology
The term "neoplasm" is a synonym of "tumor". "Neoplasia" denotes the process of the formation of neoplasms/tumors, the process is referred to as a "neoplastic" process. The word "neoplastic" itself comes from the Greek neo ("new") and plastic ("formed, molded").
The term "tumor" derives from the Latin noun tumor, "a swelling" - ultimately from the verb tumēre "to swell". In the British Commonwealth the spelling "tumour" is commonly used, whereas in the U.S. the word is usually spelled "tumor".
In its medical sense "tumor" has traditionally meant an abnormal swelling of the flesh. The Roman medical encyclopedist Celsus (c. 30 BC–38 AD) described the four cardinal signs of acute inflammation as tumor, dolor, calor, and rubor (swelling, pain, increased heat, and redness). (His treatise, De Medicina, was the first medical book printed in 1478 following the invention of the movable-type printing press.)
In contemporary English, the word "tumor" is often used as a synonym for a cystic (liquid-filled) growth or solid neoplasm (cancerous or non-cancerous),[60] with other forms of swelling often referred to as "swellings".[61]
Related terms occur commonly in the medical literature, where the nouns "tumefaction" and "tumescence" (derived from the adjective "tumescent"[62]), are current medical terms for non-neoplastic swelling. This type of swelling is most often caused by inflammation caused by trauma, infection, and other factors.
Tumors may be caused by conditions other than an overgrowth of neoplastic cells, however. Cysts (such as sebaceous cysts) are also referred to as tumors, even though they have no neoplastic cells. This is standard in medical-billing terminology (especially when billing for a growth whose pathology has yet to be determined).
See also
- Somatic evolution in cancer
- List of biological development disorders
- Epidemiology of cancer
- Pleomorphism
References
- https://www.lexico.com/en/definition/neoplasm
- Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, Delbono O (July 2014). "Type-2 pericytes participate in normal and tumoral angiogenesis". Am. J. Physiol., Cell Physiol. 307 (1): C25–38. doi:10.1152/ajpcell.00084.2014. PMC 4080181. PMID 24788248.
- Cooper GM (1992). Elements of human cancer. Boston: Jones and Bartlett Publishers. p. 16. ISBN 978-0-86720-191-8.
- Taylor, Elizabeth J. (2000). Dorland's Illustrated medical dictionary (29th ed.). Philadelphia: Saunders. p. 1184. ISBN 978-0721662541.
- Stedman's medical dictionary (28th ed.). Philadelphia: Lippincott Williams & Wilkins. 2006. p. Neoplasm. ISBN 978-0781733908.
- "II Neoplasms". International Statistical Classification of Diseases and Related Health Problems 10th Revision (ICD-10) Version for 2010. World Health Organization. Retrieved 19 June 2014.
- Abrams, Gerald. "Neoplasia I". Retrieved 23 January 2012.
- "Cancer - Activity 1 - Glossary, page 4 of 5". Archived from the original on 2008-05-09. Retrieved 2008-01-08.
- Clone definition - Medical Dictionary definitions of popular medical terms easily defined on MedTerms
- Lee ES, Locker J, Nalesnik M, Reyes J, Jaffe R, Alashari M, Nour B, Tzakis A, Dickman PS (January 1995). "The association of Epstein-Barr virus with smooth-muscle tumors occurring after organ transplantation". N. Engl. J. Med. 332 (1): 19–25. doi:10.1056/NEJM199501053320104. PMID 7990861.
- "Pancreas Cancer: Glossary of Terms". Retrieved 2008-01-08.
- "Tumor". Dorland's Illustrated Medical Dictionary (31st ed.). Saunders. 2007. ISBN 978-1-84972-348-0.
- Asashima M, Oinuma T, Meyer-Rochow VB (1987). "Tumors in amphibia". Zoological Science. 4: 411–425.
- Ambrosi D, Mollica F (2002). "On the mechanics of a growing tumor". International Journal of Engineering Science. 40 (12): 1297–316. doi:10.1016/S0020-7225(02)00014-9.
- Volokh KY (September 2006). "Stresses in growing soft tissues". Acta Biomater. 2 (5): 493–504. doi:10.1016/j.actbio.2006.04.002. PMID 16793355.
- Kastan MB (2008). "DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture". Mol. Cancer Res. 6 (4): 517–24. doi:10.1158/1541-7786.MCR-08-0020. PMID 18403632.
- Cunningham FH, Fiebelkorn S, Johnson M, Meredith C (November 2011). "A novel application of the Margin of Exposure approach: segregation of tobacco smoke toxicants". Food Chem. Toxicol. 49 (11): 2921–33. doi:10.1016/j.fct.2011.07.019. PMID 21802474.
- Kanavy HE, Gerstenblith MR (December 2011). "Ultraviolet radiation and melanoma". Semin Cutan Med Surg. 30 (4): 222–8. doi:10.1016/j.sder.2011.08.003. PMID 22123420.
- Handa O, Naito Y, Yoshikawa T (2011). "Redox biology and gastric carcinogenesis: the role of Helicobacter pylori". Redox Rep. 16 (1): 1–7. doi:10.1179/174329211X12968219310756. PMID 21605492.
- Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H (August 2011). "Carcinogenicity of deoxycholate, a secondary bile acid". Arch. Toxicol. 85 (8): 863–71. doi:10.1007/s00204-011-0648-7. PMC 3149672. PMID 21267546.
- Katsurano M, Niwa T, Yasui Y, Shigematsu Y, Yamashita S, Takeshima H, Lee MS, Kim YJ, Tanaka T, Ushijima T (January 2012). "Early-stage formation of an epigenetic field defect in a mouse colitis model, and non-essential roles of T- and B-cells in DNA methylation induction". Oncogene. 31 (3): 342–51. doi:10.1038/onc.2011.241. PMID 21685942.
- Malkin D (April 2011). "Li-fraumeni syndrome". Genes Cancer. 2 (4): 475–84. doi:10.1177/1947601911413466. PMC 3135649. PMID 21779515.
- Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K (July 2000). "Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland". N. Engl. J. Med. 343 (2): 78–85. doi:10.1056/NEJM200007133430201. PMID 10891514.
- Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I (June 2005). "O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions". Gut. 54 (6): 797–802. doi:10.1136/gut.2004.059535. PMC 1774551. PMID 15888787.
- Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JP (September 2005). "MGMT promoter methylation and field defect in sporadic colorectal cancer". J. Natl. Cancer Inst. 97 (18): 1330–8. doi:10.1093/jnci/dji275. PMID 16174854.
- Psofaki V, Kalogera C, Tzambouras N, Stephanou D, Tsianos E, Seferiadis K, Kolios G (July 2010). "Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas". World J. Gastroenterol. 16 (28): 3553–60. doi:10.3748/wjg.v16.i28.3553. PMC 2909555. PMID 20653064.
- Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Lee JH (October 2011). "Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence". Langenbecks Arch Surg. 396 (7): 1017–26. doi:10.1007/s00423-011-0812-9. PMID 21706233.
- Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, Cassingena A, Rusconi F, Esposito A, Nichelatti M, Esteller M, Siena S (April 2013). "Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer". Clin. Cancer Res. 19 (8): 2265–72. doi:10.1158/1078-0432.CCR-12-3518. PMID 23422094.
- Mokarram P, Zamani M, Kavousipour S, Naghibalhossaini F, Irajie C, Moradi Sarabi M, et al. (May 2013). "Different patterns of DNA methylation of the two distinct O6-methylguanine-DNA methyltransferase (O6-MGMT) promoter regions in colorectal cancer". Mol. Biol. Rep. 40 (5): 3851–7. doi:10.1007/s11033-012-2465-3. PMID 23271133.
- Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, et al. (May 2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology. 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, et al. (April 2010). "Modulation of mismatch repair and genomic stability by miR-155". Proc. Natl. Acad. Sci. U.S.A. 107 (15): 6982–7. doi:10.1073/pnas.1002472107. PMC 2872463. PMID 20351277.
- Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, et al. (2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integr. 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (April 1997). "Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2". Proc. Natl. Acad. Sci. U.S.A. 94 (7): 3122–7. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (December 2006). "Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6". Carcinogenesis. 27 (12): 2402–8. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (March 2002). "Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation". EMBO Rep. 3 (3): 255–60. doi:10.1093/embo-reports/kvf037. PMC 1084010. PMID 11850397.
- O'Hagan HM, Mohammad HP, Baylin SB (2008). Lee JT (ed.). "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLoS Genet. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV (July 2007). "DNA damage, homology-directed repair, and DNA methylation". PLoS Genet. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. (March 2012). "Intratumor heterogeneity and branched evolution revealed by multiregion sequencing". N. Engl. J. Med. 366 (10): 883–92. doi:10.1056/NEJMoa1113205. PMC 4878653. PMID 22397650.
- Slaughter DP, Southwick HW, Smejkal W (September 1953). "Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin". Cancer. 6 (5): 963–8. doi:10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q. PMID 13094644.
- Bernstein C, Bernstein H, Payne CM, Dvorak K, Garewal H (February 2008). "Field defects in progression to gastrointestinal tract cancers". Cancer Lett. 260 (1–2): 1–10. doi:10.1016/j.canlet.2007.11.027. PMC 2744582. PMID 18164807.
- Nguyen H, Loustaunau C, Facista A, Ramsey L, Hassounah N, Taylor H, Krouse R, Payne CM, Tsikitis VL, Goldschmid S, Banerjee B, Perini RF, Bernstein C (2010). "Deficient Pms2, ERCC1, Ku86, CcOI in field defects during progression to colon cancer". J Vis Exp (41): 1931. doi:10.3791/1931. PMC 3149991. PMID 20689513.
- Rubin H (March 2011). "Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture". BioEssays. 33 (3): 224–31. doi:10.1002/bies.201000067. PMID 21254148.
- Tsao JL, Yatabe Y, Salovaara R, Järvinen HJ, Mecklin JP, Aaltonen LA, Tavaré S, Shibata D (February 2000). "Genetic reconstruction of individual colorectal tumor histories". Proc. Natl. Acad. Sci. U.S.A. 97 (3): 1236–41. doi:10.1073/pnas.97.3.1236. PMC 15581. PMID 10655514.
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (March 2013). "Cancer genome landscapes". Science. 339 (6127): 1546–58. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- Lochhead P, Chan AT, Nishihara R, Fuchs CS, Beck AH, Giovannucci E, Ogino S (2014). "Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression". Mod Pathol. 28 (1): 14–29. doi:10.1038/modpathol.2014.81. PMC 4265316. PMID 24925058.
- Svrcek M, Buhard O, Colas C, Coulet F, Dumont S, Massaoudi I, et al. (November 2010). "Methylation tolerance due to an O6-methylguanine DNA methyltransferase (MGMT) field defect in the colonic mucosa: an initiating step in the development of mismatch repair-deficient colorectal cancers". Gut. 59 (11): 1516–26. doi:10.1136/gut.2009.194787. PMID 20947886.
- Paluszczak J, Misiak P, Wierzbicka M, Woźniak A, Baer-Dubowska W (February 2011). "Frequent hypermethylation of DAPK, RARbeta, MGMT, RASSF1A and FHIT in laryngeal squamous cell carcinomas and adjacent normal mucosa". Oral Oncol. 47 (2): 104–7. doi:10.1016/j.oraloncology.2010.11.006. PMID 21147548.
- Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA, Smoller BR, Kokoska MS, Fan CY (October 2009). "Increased microsatellite instability and epigenetic inactivation of the hMLH1 gene in head and neck squamous cell carcinoma". Otolaryngol Head Neck Surg. 141 (4): 484–90. doi:10.1016/j.otohns.2009.07.007. PMID 19786217.
- Tawfik HM, El-Maqsoud NM, Hak BH, El-Sherbiny YM (2011). "Head and neck squamous cell carcinoma: mismatch repair immunohistochemistry and promoter hypermethylation of hMLH1 gene". Am J Otolaryngol. 32 (6): 528–36. doi:10.1016/j.amjoto.2010.11.005. PMID 21353335.
- Zou XP, Zhang B, Zhang XQ, Chen M, Cao J, Liu WJ (November 2009). "Promoter hypermethylation of multiple genes in early gastric adenocarcinoma and precancerous lesions". Hum. Pathol. 40 (11): 1534–42. doi:10.1016/j.humpath.2009.01.029. PMID 19695681.
- Wani M, Afroze D, Makhdoomi M, Hamid I, Wani B, Bhat G, Wani R, Wani K (2012). "Promoter methylation status of DNA repair gene (hMLH1) in gastric carcinoma patients of the Kashmir valley". Asian Pac. J. Cancer Prev. 13 (8): 4177–81. doi:10.7314/APJCP.2012.13.8.4177. PMID 23098428.
- Agarwal A, Polineni R, Hussein Z, Vigoda I, Bhagat TD, Bhattacharyya S, Maitra A, Verma A (2012). "Role of epigenetic alterations in the pathogenesis of Barrett's esophagus and esophageal adenocarcinoma". Int J Clin Exp Pathol. 5 (5): 382–96. PMC 3396065. PMID 22808291.
- Hofstad B, Vatn MH, Andersen SN, Huitfeldt HS, Rognum T, Larsen S, Osnes M (September 1996). "Growth of colorectal polyps: redetection and evaluation of unresected polyps for a period of three years". Gut. 39 (3): 449–56. doi:10.1136/gut.39.3.449. PMC 1383355. PMID 8949653.
- Schmitt MW, Prindle MJ, Loeb LA (September 2012). "Implications of genetic heterogeneity in cancer". Ann. N. Y. Acad. Sci. 1267: 110–6. doi:10.1111/j.1749-6632.2012.06590.x. PMC 3674777. PMID 22954224.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. (February 2001). "Initial sequencing and analysis of the human genome" (PDF). Nature. 409 (6822): 860–921. doi:10.1038/35057062. PMID 11237011.
- Yost SE, Smith EN, Schwab RB, Bao L, Jung H, Wang X, Voest E, Pierce JP, Messer K, Parker BA, Harismendy O, Frazer KA (August 2012). "Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens". Nucleic Acids Res. 40 (14): e107. doi:10.1093/nar/gks299. PMC 3413110. PMID 22492626.
- Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. (May 2012). "Melanoma genome sequencing reveals frequent PREX2 mutations". Nature. 485 (7399): 502–6. doi:10.1038/nature11071. PMC 3367798. PMID 22622578.
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, et al. (April 2010). "Analysis of genetic inheritance in a family quartet by whole-genome sequencing". Science. 328 (5978): 636–9. doi:10.1126/science.1186802. PMC 3037280. PMID 20220176.
- Campbell CD, Chong JX, Malig M, Ko A, Dumont BL, Han L, et al. (November 2012). "Estimating the human mutation rate using autozygosity in a founder population". Nat. Genet. 44 (11): 1277–81. doi:10.1038/ng.2418. PMC 3483378. PMID 23001126.
- Tumor in Medical Encyclopedia
- "Swelling". MedlinePlus Medical Encyclopedia. October 14, 2012.
- "tumescence". Oxford English Dictionary (3rd ed.). Oxford University Press. September 2005. (Subscription or UK public library membership required.)
External links
| Classification | |
|---|---|
| External resources |