Frontotemporal lobar degeneration
Frontotemporal lobar degeneration (FTLD) is a pathological process that occurs in frontotemporal dementia. It is characterized by atrophy in the frontal lobe and temporal lobe of the brain, with sparing of the parietal and occipital lobes.
| Frontotemporal lobar degeneration | |
|---|---|
| Specialty | Psychiatry, neurology |
Common proteinopathies that are found in FTLD include the accumulation of tau proteins and TARDBPs. Mutations in the C9orf72 gene have been established as a major genetic contribution of FTLD, although defects in the GRN and MAPT genes are also associated with it.[1]
Classification
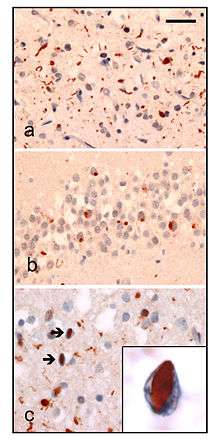
There are 3 main histological subtypes found at post-mortem:
- FTLD-tau is characterised by tau positive inclusions often referred to as Pick-bodies. Examples of FTLD-tau include; Pick's disease, corticobasal degeneration, progressive supranuclear palsy.
- FTLD-TDP (or FTLD-U ) is characterised by ubiquitin and TDP-43 positive, tau negative, FUS negative inclusions. The pathological histology of this subtype is so diverse it is subdivided into four subtypes based on the detailed histological findings:
- Type A presents with many small neurites and neuronal cytoplasmic inclusions in the upper (superficial) cortical layers. Bar-like neuronal intranuclear inclusions can also be seen they are fewer in number.
- Type B presents with lots of neuronal and glial cytoplasmic inclusions in both the upper (superficial) and lower (deep) cortical layers, and lower motor neurons. However neuronal intranuclear inclusions are rare or absent. This is often associated with ALS and C9ORF72 mutations (see next section).
- Type C presents many long neuritic profiles found in the superficial cortical laminae, very few or no neuronal cytoplasmic inclusions, neuronal intranuclear inclusions or glial cytoplasmic inclusions. This is often associated with semantic dementia.
- Type D presents with lots of neuronal intranuclear inclusions and dystrophic neurites, and an unusual absence of inclusions in the granual cell layer of the hippocampus. Type 4 is associated with VCP mutations.
Two physicians independently categorized the various forms of TDP-43 associated disorders. Both classifications were considered equally valid by the medical community, but the physicians in question have jointly proposed a compromise classification to avoid confusion.[2]
- FTLD-FUS; which is characterised by FUS positive cytoplasmic inclusions, intra nuclear inclusions, and neuritic threads. All of which are present in the cortex, medulla, hippocampus, and motor cells of the spinal cord and XIIth cranial nerve.
Dementia lacking distinctive histology (DLDH) is a rare and controversial entity. New analyses have allowed many cases previously described as DLDH to be reclassified into one of the positively defined subgroups.
Genetics
There have been numerous advances in descriptions of genetic causes of FTLD, and the related disease amyotrophic lateral sclerosis.
- Mutations in the Tau gene (known as MAPT or Microtubule Associated Protein Tau) can cause a FTLD presenting with tau pathology (FTLD-tau).[3] There are over 40 known mutations at present.
- Mutations in the Progranulin gene (PGRN) can cause a FTLD presenting with TDP-43 pathology (FTLD-TDP43). Patients with Progranulin mutations have type 3 ubiquitin-positive, TDP-43 positive, tau-negative pathology at post-mortem. Progranulin is associated with tumorgenesis when overproduced, however the mutations seen in FTLD-TDP43 produce a haploinsufficiency, meaning that because one of the two alleles is damaged, only half as much Progranulin is produced.[4]
- Mutations in the CHMP2B gene are associated with a rare behavioural syndrome akin to bvFTLD (mainly in a large Jutland cohort), presenting with a tau negative, TDP-43 negative, FUS negative, Ubiquitin positive pathology.
- Mutations in the VCP have been shown to cause a TDP-43 positive FTLD which is associated with the IBMPFD syndrome (inclusion body myopathy, Paget's disease and frontotemporal dementia)[5]
- Mutations in the TDP-43 gene (known as TARBP or TAR DNA-binding protein) are an exceptionally rare cause of FTLD, despite this protein being present in the pathological inclusions of many cases (FTLD-TDP43).[6] However, mutations in TARBP are a more common cause of ALS, which can present with frontotemporal dementia. Since these instances are not considered a pure FTLD they are not included here.
Mutations in all of the above genes cause a very small fraction of the FTLD spectrum. Most of the cases are sporadic (no known genetic cause).
- A proportion of FTLD-TDP43 [with ALS] cases had shown genetic linkage to a region on chromosome 9 (FTLD-TDP43/Ch9). This linkage has recently been pinned down to the C9ORF72 gene. Two groups published identical findings back-to-back in the journal Neuron in mid-2011, showing that a hexanucleotide repeat expansion of the GGGGCC genetic sequence within an intron of this gene was responsible. This expansion was found to be present in a large proportion of familial and sporadic cases, particularly in the Finnish population[7]
Diagnosis
For diagnostic purposes, magnetic resonance imaging (MRI) and ([18F]fluorodeoxyglucose) positron emission tomography (FDG-PET) are applied. They measure either atrophy or reductions in glucose utilization. The three clinical subtypes of frontotemporal lobar degeneration, frontotemporal dementia, semantic dementia and progressive nonfluent aphasia, are characterized by impairments in specific neural networks.[8] The first subtype with behavioral deficits, frontotemporal dementia, mainly affects a frontomedian network discussed in the context of social cognition. Semantic dementia is mainly related to the inferior temporal poles and amygdalae; brain regions that have been discussed in the context of conceptual knowledge, semantic information processing, and social cognition, whereas progressive nonfluent aphasia affects the whole left frontotemporal network for phonological and syntactical processing.
Examples
United States Senator Pete Domenici (R-NM) was a known sufferer of FTLD, and the illness was the main reason behind his October 4, 2007 announcement of retirement at the end of his term. American film director, producer, and screenwriter Curtis Hanson died as a result of FTLD on September 20, 2016.
References
- van der Zee, Julie; Van Broeckhoven, Christine (7 January 2014). "Dementia in 2013: Frontotemporal lobar degeneration—building on breakthroughs". Nature Reviews Neurology. 10 (2): 70–72. doi:10.1038/nrneurol.2013.270. PMID 24394289.
- Ian R. A. Mackenzie; Manuela Neumann; Atik Baborie; Deepak M. Sampathu; Daniel Du Plessis; Evelyn Jaros; Robert H. Perry; John Q. Trojanowski; David M. A. Mann & Virginia M. Y. Lee (July 2011). "A harmonized classification system for FTLD-TDP pathology". Acta Neuropathol. 122 (1): 111–113. doi:10.1007/s00401-011-0845-8. PMC 3285143. PMID 21644037.
- Goedert, M.; et al. (1989). "Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain". The EMBO Journal. 8 (2): 393–9. doi:10.1002/j.1460-2075.1989.tb03390.x. PMC 400819. PMID 2498079.
- Cruts, M.; et al. (2006). "Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21". Nature. 442 (7105): 920–4. Bibcode:2006Natur.442..920C. doi:10.1038/nature05017. PMID 16862115.
- Kimonis, V.E.; et al. (2008). "VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder" (PDF). Biochim Biophys Acta. 1782 (12): 744–8. doi:10.1016/j.bbadis.2008.09.003. PMID 18845250.
- Borroni, B.; et al. (2010). "TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course". Rejuvenation Res. 13 (5): 509–17. doi:10.1089/rej.2010.1017. PMID 20645878.
- Dejesus-Hernandez, M.; et al. (2011). "Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS". Neuron. 72 (2): 245–56. doi:10.1016/j.neuron.2011.09.011. PMC 3202986. PMID 21944778.
- Schroeter ML, Raczka KK, Neumann J, von Cramon DY (2007). "Towards a nosology for frontotemporal lobar degenerations – A meta-analysis involving 267 subjects". NeuroImage. 36 (3): 497–510. doi:10.1016/j.neuroimage.2007.03.024. PMID 17478101.
Bibliography
- Mackenzie IR, Baborie A, Pickering-Brown S, et al. (November 2006). "Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype". Acta Neuropathologica. 112 (5): 539–49. doi:10.1007/s00401-006-0138-9. PMC 2668618. PMID 17021754.
- Davidson Y, Kelley T, Mackenzie IR, et al. (May 2007). "Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43". Acta Neuropathologica. 113 (5): 521–33. doi:10.1007/s00401-006-0189-y. PMID 17219193.
- Neary D, Snowden JS, Gustafson L, et al. (December 1, 1998). "Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria". Neurology. 51 (6): 1546–54. doi:10.1212/wnl.51.6.1546. PMID 9855500. Retrieved 2009-06-20.
- Pickering-Brown SM (July 2007). "The complex aetiology of frontotemporal lobar degeneration". Experimental Neurology. 206 (1): 1–10. doi:10.1016/j.expneurol.2007.03.017. PMID 17509568.
Further reading
- Hodges, John R. The Frontotemporal Dementia Syndromes. Cambridge University Press. 2007 ISBN 978-0-521-85477-1