Bardet–Biedl syndrome
Bardet–Biedl syndrome (BBS) is a ciliopathic human genetic disorder that produces many effects and affects many body systems. It is characterized principally by obesity, retinitis pigmentosa, polydactyly, hypogonadism, and kidney failure in some cases.[2] Historically, slower mental processing has also been considered a principal symptom but is now not regarded as such.
- Laurence-Moon-Biedl syndrome and Laurence-Moon-Biedl-Bardet redirect here. See below for an explanation.
| Bardet–Biedl syndrome | |
|---|---|
| Other names | Biedl-Bardet Syndrome [1] |
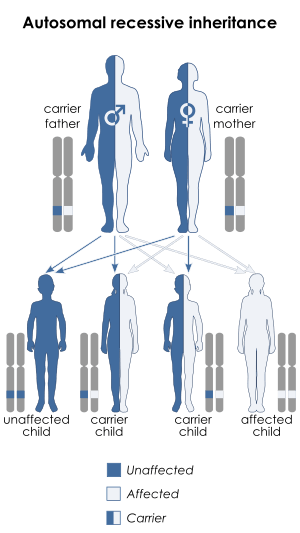 | |
| This condition can be inherited via autosomal recessive manner(including digenic recessive ) | |
| Specialty | Medical genetics |
Signs and symptoms
Bardet–Biedl syndrome is a pleiotropic disorder with variable expressivity and a wide range of clinical variability observed both within and between families. The main clinical features are rod–cone dystrophy, with childhood-onset visual loss preceded by night blindness; postaxial polydactyly; truncal obesity that manifests during infancy and remains problematic throughout adulthood; specific learning difficulties in some but not all individuals; male hypogenitalism and complex female genitourinary malformations; and renal dysfunction, a major cause of morbidity and mortality.
There is a wide range of secondary features that are sometimes associated with BBS[3] including[4]
- Strabismus, cataracts, astigmatism, pigmentary retinopathy, poor visual acuity, low vision, and/or blindness caused by an impaired photoreceptor transport mechanism in the retina.[5]
- "Brachydactyly, syndactyly of both the hands and feet is common, as is partial syndactyl (most usually between the second and third toes)"
- Polyuria/polydipsia (nephrogenic diabetes insipidus)
- Ataxia/poor coordination/imbalance
- Mild hypertonia (especially lower limbs)
- Diabetes mellitus
- Hepatic involvement
- Anosmia
- Auditory deficiencies
- Hirschsprung disease and subsequent bowel obstruction has been described.[6]
- Hypertrophy of interventricular septum and left ventricle and dilated cardiomyopathy.
- Hypogonadism, renal failure, urogenital sinuses, ectopic urethra, uterus duplex, septate vagina, and hypoplasia of the uterus, ovaries, and fallopian tubes
- Speech disorder/delay
- Developmental delay, especially of fine and gross motor skills
Relation to other rare genetic disorders
Recent findings in genetic research have suggested that a large number of genetic disorders, both genetic syndromes and genetic diseases, that were not previously identified in the medical literature as related, may be, in fact, highly related in the genetypical root cause of the widely varying, phenotypically observed disorders. BBS is one such syndrome that has now been identified to be caused by defects in the cellular ciliary structure. Thus, BBS is a ciliopathy. Other known ciliopathies include primary ciliary dyskinesia, polycystic kidney and liver disease, nephronophthisis, Alström syndrome, Meckel–Gruber syndrome and some forms of retinal degeneration.[7]
Pathophysiology
The detailed biochemical mechanism that leads to BBS is still unclear.
The gene products encoded by these BBS genes, called BBS proteins, are located in the basal body and cilia of the cell.[8]
Using the round worm C. elegans as a model system, biologists found that BBS proteins are involved in a process called Intraflagellar transport (IFT), a bi-directional transportation activity within the cilia along the long axis of the ciliary shaft that is essential for ciliogenesis and the maintenance of cilia.[9] Recent biochemical analysis of human BBS proteins revealed that BBS proteins are assembled into a multiple protein complex, called "BBSome". BBSome is proposed to be responsible for transporting intracellular vesicles to the base of the cilia and to play an important role in the ciliary function.
Since abnormalities of cilia are known to be related to a wide range of disease symptoms including those commonly seen in BBS patients, it is now widely accepted that mutated BBS genes affect normal cilia function, which, in turns, causes BBS.
A theory that photoreceptor cells are nourished by the IFT of retinal cilia now offers a potential explanation for the retinal dystrophy common in BBS patients after their early years of life.[10][11]
Genes involved include:
- BBsome: BBS1, BBS2, ARL6/BBS3, BBS4, BBS5, BBS7, TTC8/BBS8, BBS10, TRIM32/BBS11 BBS12, CCDC28B, CEP290, TMEM67, MKS1, MKKS[12]
- chaperone: BBS6
Diagnosis
The diagnosis of BBS is established by clinical findings and family history. Molecular genetic testing can be used to confirm the diagnosis. Multigene panels offer the most effective approach in achieving molecular confirmation of BBS.
Eponym
The syndrome is named after Georges Bardet and Arthur Biedl.[13] The first known case was reported by Laurence and Moon in 1866 at the Ophthalmic Hospital in South London. Laurence–Moon–Biedl–Bardet syndrome is no longer considered as valid terms in that patients of Laurence and Moon had paraplegia but no polydactyly or obesity, which are the key elements of the Bardet–Biedl syndrome. Laurence–Moon syndrome is usually considered a separate entity. However, some recent research suggests that the two conditions may not be distinct.[14]
As of 2012, 14[12] (or 15)[15] different BBS genes had been identified.
References
- "Bardet-Biedl syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 13 August 2019.
- Beales P, Elcioglu N, Woolf A, Parker D, Flinter F (1 June 1999). "New criteria for improved diagnosis of Bardet–Biedl syndrome: results of a population survey". J. Med. Genet. 36 (6): 437–46. doi:10.1136/jmg.36.6.437 (inactive 2019-08-25). PMC 1734378. PMID 10874630. Archived from the original on 2008-03-14. Retrieved 2007-10-11.
- Ross, Allison; PL Beales; J Hill (2008). The Clinical, Molecular, and Functional Genetics of Bardet–Biedl Syndrome, in Genetics of Obesity Syndromes. Oxford University Press. pp. 147–148. ISBN 978-0-19-530016-1. Retrieved 2009-07-01.
- Ross, Allison; PL Beales; J Hill (2008). The Clinical, Molecular, and Functional Genetics of Bardet–Biedl Syndrome, in Genetics of Obesity Syndromes. Oxford University Press. pp. 153–154. ISBN 978-0-19-530016-1. Retrieved 2009-07-01.
- Abd-El-Barr, MM; Sykoudis K; Andrabi S; Eichers ER; Pennesi ME; Tan PL; Wilson JH; Katsanis N; Lupski JR; Wu SM. (December 2007). "Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet–Biedl syndrome". Vision Res. 47 (27): 3394–407. doi:10.1016/j.visres.2007.09.016. PMC 2661240. PMID 18022666.
- Ramji AN. Sigmoid volvulus in bardet-biedl syndrome: serendipity or a new association? Int Surg J 2019;6:1388-91.
- Badano JL, Mitsuma N, Beales PL, Katsanis N (2006). "The ciliopathies: an emerging class of human genetic disorders". Annu. Rev. Genom. Hum. Genet. 7: 125–48. doi:10.1146/annurev.genom.7.080505.115610. PMID 16722803.
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, Mah AK, Johnsen RC, Cavender JC, Lewis RA, Leroux MR, Beales PL, Katsanis N (October 2003). "Basal body dysfunction is a likely cause of pleiotropic Bardet–Biedl syndrome". Nature. 425 (6958): 628–33. Bibcode:2003Natur.425..628A. doi:10.1038/nature02030. PMID 14520415.
- Blacque OE, Reardon MJ, Li C, McCarthy J, Mahjoub MR, Ansley SJ, Badano JL, Mah AK, Beales PL, Davidson WS, Johnsen RC, Audeh M, Plasterk RH, Baillie DL, Katsanis N, Quarmby LM, Wicks SR, Leroux MR (2004). "Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport". Genes Dev. 18 (13): 1630–42. doi:10.1101/gad.1194004. PMC 443524. PMID 15231740.
- Sedmak T, Wolfrum U (April 2010). "Intraflagellar transport molecules in ciliary and nonciliary cells of the retina". J. Cell Biol. 189 (1): 171–86. doi:10.1083/jcb.200911095. PMC 2854383. PMID 20368623.
- Orozco, JT; Wedaman KP; Signor D; Brown H; Rose L; Scholey JM (1999). "Movement of motor and cargo along cilia". Nature. 398 (6729): 674. Bibcode:1999Natur.398..674O. doi:10.1038/19448. PMID 10227290.
- Hamosh, Ada (2012-11-02). "OMIM entry #209900 Bardet-Biedl Syndrome; BBS". Online Mendelian Inheritance in Man. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine. Archived from the original on 2016-05-17. Retrieved 2013-09-04.
- synd/3745 at Who Named It?
- Moore S, Green J, Fan Y, et al. (2005). "Clinical and genetic epidemiology of Bardet–Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study". Am. J. Med. Genet. 132 (4): 352–60. doi:10.1002/ajmg.a.30406. PMC 3295827. PMID 15637713.
- Hereditary Retinopathies: Progress in Development of Genetic and Molecular Therapies. Springer. 2012. p. 15. ISBN 9781461444992. Retrieved 2013-09-04.
External links
| Classification | |
|---|---|
| External resources |
|
- Overview at United States National Library of Medicine
- Molecular diagnosis at NCBI