Wilms' tumor
Wilms' tumor, also known as nephroblastoma, is a cancer of the kidneys that typically occurs in children, rarely in adults.[1] It is named after Max Wilms, the German surgeon (1867–1918) who first described it.[2]
| Wilms' tumor | |
|---|---|
| Other names | Wilms's tumor |
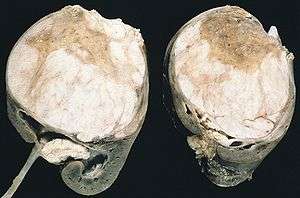 | |
| Cut section showing two halves of a nephroblastoma specimen. Note the prominent septa subdividing the sectioned surface and the protrusion of tumor into the renal pelvis, resembling botryoid rhabdomyosarcoma. | |
| Pronunciation |
|
| Specialty | Oncology, urology, nephrology |
Approximately 650 cases are diagnosed in the U.S. annually.[3] The majority of cases occur in children with no associated genetic syndromes; however, a minority of children with Wilms' tumor have a congenital abnormality.[3] It is highly responsive to treatment, with about 9/10 children being cured.[3]
Signs and symptoms
Typical signs and symptoms of Wilms' tumor include the following:
- a painless, palpable abdominal mass
- loss of appetite
- abdominal pain
- fever
- nausea and vomiting
- blood in the urine (in about 20% of cases)
- high blood pressure in some cases (especially if synchronous or metachronous bilateral kidney involvement)
- Rarely as varicocele[4]
Pathogenesis
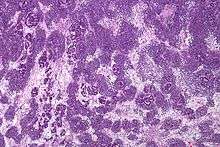
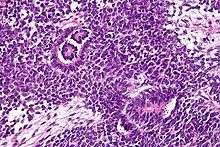
Wilms' tumor has many causes, which can broadly be categorized as syndromic and non-syndromic. Syndromic causes of Wilms' tumor occur as a result of alterations to genes such as the Wilms Tumor 1 (WT1) or Wilms Tumor 2 (WT2) genes, and the tumor presents with a group of other signs and symptoms.[5] Non-syndromic Wilms' tumor is not associated with other symptoms or pathologies.[5] Many, but not all, cases of Wilms' tumor develop from nephrogenic rests, which are fragments of tissue in or around the kidney that develop before birth and become cancerous after birth. In particular, cases of bilateral Wilms' tumor, as well as cases of Wilms' tumor derived from certain genetic syndromes such as Denys-Drash syndrome, are strongly associated with nephrogenic rests.[5] Most nephroblastomas are on one side of the body only and are found on both sides in less than 5% of cases, although people with Denys-Drash syndrome mostly have bilateral or multiple tumors.[6] They tend to be encapsulated and vascularized tumors that do not cross the midline of the abdomen. In cases of metastasis it is usually to the lung. A rupture of Wilms' tumor puts the patient at risk of bleeding and peritoneal dissemination of the tumor. In such cases, surgical intervention by a surgeon who is experienced in the removal of such a fragile tumor is imperative.
Pathologically, a triphasic nephroblastoma comprises three elements:
- blastema
- mesenchyme (stroma)
- epithelium
Wilms' tumor is a malignant tumor containing metanephric blastema, stromal and epithelial derivatives. Characteristic is the presence of abortive tubules and glomeruli surrounded by a spindled cell stroma. The stroma may include striated muscle, cartilage, bone, fat tissue, and fibrous tissue. Dysfunction is caused when the tumor compresses the normal kidney parenchyma.
The mesenchymal component may include cells showing rhabdomyoid differentiation or malignancy (rhabdomyosarcomatous Wilms).
Wilms' tumors may be separated into 2 prognostic groups based on pathologic characteristics:
- Favorable - Contains well developed components mentioned above
- Anaplastic - Contains diffuse anaplasia (poorly developed cells)
Molecular biology and related conditions
Mutations of the WT1 gene on chromosome 11p13 are observed in approximately 20% of Wilms' tumors.[7][8] At least half of the Wilms' tumors with mutations in WT1 also carry mutations in CTNNB1, the gene encoding the proto-oncogene beta-catenin.[9]
Most cases do not have mutations in any of these genes.[10]
| Syndrome Name | Associated Genetic Variant | Risk for Wilms tumor | Description of Syndrome |
| WAGR syndrome (Wilms tumor, aniridia, genital anomalies, retardation) | Gene deletion that includes both WT1 and PAX6 | 45-60% | Characterized by Wilms tumor, aniridia (absence of iris), hemihypertrophy (one side of body larger than the other), genitourinary abnormalities, ambiguous genitalia, intellectual disability.[11] |
| Denys-Drash syndrome (DDS) | WT1 (exon 8 and 9) | 74% | Characterized by kidney diseases since birth leading to early-onset kidney failure, ambiguous genitalia (intersex disorders).[11] |
| Beckwith-Wiedemann Syndrome | Abnormal regulation of chromosome 11p15.5 | 7% | Characterized by macrosmia (large birth size), macroglossia (large tongue), hemihypertrophy (one side of the body is larger), other tumors in body, omphalocele (open abdominal wall) and visceromegaly (enlargement of organs inside abdomen).[11] |
Diagnosis
The majority of people with Wilms’ tumor present with an asymptotic abdominal mass which is noticed by a family member or healthcare professional.[12] Renal tumors can also be found during routine screening in children who have known predisposing clinical syndromes.[12] The diagnostic process includes taking a medical history, a physical exam, and a series of tests including blood, urine, and imaging tests.[13]
Once Wilms’ tumor is suspected, an ultrasound scan is usually done first to confirm the presence of an intrarenal mass.[13] A computed tomography scan or MRI scan can also be used for more detailed imaging. Finally, the diagnosis of Wilms’ tumor is confirmed by a tissue sample.[14] In most cases, a biopsy is not done first because there is a risk of cancer cells spreading during the procedure. Treatment in North America is nephrectomy or in Europe chemotherapy followed by nephrectomy. A definitive diagnosis is obtained by pathological examination of the nephrectomy specimen.[14]
Staging
Staging is a standard way to describe the extent of spread of Wilms' tumors,[15] and to determine prognosis and treatments. Staging is based on anatomical findings and tumor cells pathology.[16][17] According to the extend of tumor tissue at the time of initial diagnosis, five stages are considered.
In Stage I Wilms' tumor (43% of cases), all of the following criteria must be met:
- Tumor is limited to the kidney and is completely excised.
- The surface of the renal capsule is intact.
- The tumor is not ruptured or biopsied (open or needle) prior to removal.
- No involvement of extrarenal or renal sinus lymph-vascular spaces
- No residual tumor apparent beyond the margins of excision.
- Metastasis of tumor to lymph nodes not identified.
In Stage II (23% of cases), 1 or more of the following criteria must be met:
- Tumor extends beyond the kidney but is completely excised.
- No residual tumor apparent at or beyond the margins of excision.
- Any of the following conditions may also exist:
- Tumor involvement of the blood vessels of the renal sinus and/or outside the renal parenchyma.
- Extensive tumor involvement of renal sinus soft tissue.
In Stage III (20% of cases), 1 or more of the following criteria must be met:
- Inoperable primary tumor.
- Lymph node metastasis.
- Tumor is present at surgical margins.
- Tumor spillage involving peritoneal surfaces either before or during surgery, or transected tumor thrombus.
- The tumor has been biopsied prior to removal or there is local spillage of tumor during surgery, confined to the flank.
Stage IV (10% of cases) Wilms' tumor is defined by the presence of hematogenous metastases (lung, liver, bone, or brain), or lymph node metastases outside the abdomenopelvic region.
Stage V (5% of cases) Wilms' tumor is defined by bilateral renal involvement at the time of initial diagnosis. For patients with bilateral involvement, an attempt should be made to stage each side according to the above criteria (stage I to III) on the basis of extent of disease prior to biopsy.
Treatment/prognosis
The overall 5-year survival is estimated to be approximately 90%,[18][19] but for individuals the prognosis is highly dependent on individual staging and treatment. Early removal tends to promote positive outcomes.
Tumor-specific loss-of-heterozygosity (LOH) for chromosomes 1p and 16q identifies a subset of Wilms' tumor patients who have a significantly increased risk of relapse and death. LOH for these chromosomal regions can now be used as an independent prognostic factor together with disease stage to target intensity of treatment to risk of treatment failure.[20][21] Genome-wide copy number and LOH status can be assessed with virtual karyotyping of tumor cells (fresh or paraffin-embedded).
Statistics may sometimes show more favorable outcomes for more aggressive stages than for less aggressive stages, which may be caused by more aggressive treatment and/or random variability in the study groups. Also, a stage V tumor is not necessarily worse than a stage IV tumor.
| Stage[22] | Histopathology[22] | 4 Year relapse-free survival (RFS) or event-free survival (EFS)[22] | 4 Year overall survival (OS)[22] | Treatment[22] |
|---|---|---|---|---|
| Stage I[22] | Favorable histology in children younger than 24 months or tumor weight less than 550g | 85% | 98% | Surgery only (should be done only within the context of a clinical trial) |
| Favorable histology in children older than 24 months or tumor weight more than 550g | 94% RFS | 98% | Nephrectomy + lymph node sampling followed by regimen EE-4A | |
| Diffuse anaplastic | 68% EFS | 80% | Nephrectomy + lymph node sampling followed by regimen EE-4A and radiotherapy | |
| Stage II[22] | Favorable histology | 86% RFS | 98% | Nephrectomy + lymph node sampling followed by regimen EE-4A |
| Focal anaplastic | 80% EFS | 80% | Nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen DD-4A | |
| Diffuse anaplastic | 83% EFS | 82% | Nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen I | |
| Stage III[22] | Favorable histology | 87% RFS | 94% | Nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen DD-4A |
| Focal anaplastic | 88% RFS | 100% (8 people in study) | Nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen DD-4A | |
| Focal anaplastic (preoperative treatment) | 71% RFS | 71% | Preoperative treatment with regimen DD-4A followed by nephrectomy + lymph node sampling and abdominal radiotherapy | |
| Diffuse anaplastic | 46% EFS | 53% | Preoperative treatment with regimen I followed by nephrectomy + lymph node sampling and abdominal radiotherapy | |
| Diffuse anaplastic | 65% EFS | 67% | Immediate nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen I | |
| Stage IV[22] | Favorable histology | 76% RFS | 86% | Nephrectomy + lymph node sampling, followed by abdominal radiotherapy, bilateral pulmonary radiotherapy, and regimen DD-4A |
| Focal anaplastic | 61% EFS | 72% | Nephrectomy + lymph node sampling, followed by abdominal radiotherapy, bilateral pulmonary radiotherapy, and regimen DD-4A | |
| Diffuse anaplastic | 33% EFS | 33% | Immediate nephrectomy + lymph node sampling followed by abdominal radiotherapy, whole-lung radiotherapy, and regimen I | |
| Diffuse anaplastic (preoperative treatment) | 31% EFS | 44% | Preoperative treatment with regimen I followed by nephrectomy + lymph node sampling followed by abdominal radiotherapy, whole-lung radiotherapy | |
| Stage V[22] | Overall | 61% EFS | 80% | |
| Favorable histology | 65% | 87% | Preoperative treatment with regimen DD-4A, followed by nephron sparing surgery or nephrecomy, staging of tumors, and chemotherapy and/or radiotherapy based on pathology and staging | |
| Focal anaplastic | 76% | 88% | Preoperative treatment with regimen DD-4A, followed by nephron sparing surgery or nephrecomy, staging of tumors, and chemotherapy and/or radiotherapy based on pathology and staging | |
| Diffuse anaplastic | 25% | 42% | Preoperative treatment with regimen DD-4A, followed by nephron sparing surgery or nephrecomy, staging of tumors, and chemotherapy and/or radiotherapy based on pathology and staging |
In case of relapse of Wilms' tumor, the 4-year survival rate for children with a standard-risk has been estimated to be 80%.[23]
Epidemiology
Wilms tumor is the most common malignant renal tumor in children.[24] There are a number of rare genetic syndromes that have been linked to an increased risk of developing Wilms Tumor.[25] Screening guidelines vary between countries; however health care professionals are recommending regular ultrasound screening for people with associated genetic syndromes.[25]
Wilms' tumor affects approximately one person per 10,000 worldwide before the age of 15 years.[26] People of African descent may have slightly higher rates of Wilms' tumor.[26] The peak age of Wilms' tumor is 3 to 4 years and most cases occur before the age of 10 years.[27] A genetic predisposition to Wilms' tumor in individuals with aniridia has been established, due to deletions in the p13 band on chromosome 11.[28]
History
Dr. Sidney Farber, founder of Dana–Farber Cancer Institute, and his colleagues achieved the first remissions in Wilms' tumor in the 1950s. By employing the antibiotic actinomycin D in addition to surgery and radiation therapy, they boosted cure rates from 40 to 89 percent.
The use of computed tomography scan for the diagnosis of Wilms' tumor began in early 1970s, thanks to the intuition of Dr.Mario Costici, an Italian physician. He discovered that in the direct radiograms and in the urographic images, you can identify determining elements for a differential diagnosis with the Wilms' tumor. This possibility was a premise for starting a treatment.[29]
Notable cases
Vince Neil's daughter Skylar died from this cancer in August 1995, a month before he released his Carved in Stone, which includes the tribute track "Skylar's Song".
See also
- Hemihypertrophy
- National Wilms Tumor Study Group (NWTS)
- Virtual Karyotype for 1p and 16q LOH
- Perlman syndrome
References
- EBSCO database verified by URAC; accessed from Mount Sinai Hospital, New York
- WhoNamedIt.com: Max Wilms
- "Wilms Tumor and Other Childhood Kidney Tumors Treatment". National Cancer Institute. Retrieved 2018-11-12.
- Williams V, Munikoty V, Trehan A (July 2018). "Varicocele with an Abdominal Mass". The Journal of Pediatrics. 198: 317–317.e1. doi:10.1016/j.jpeds.2018.02.023. PMID 29580678.
- PDQ Pediatric Treatment Editorial Board (2002), "Wilms Tumor and Other Childhood Kidney Tumors Treatment (PDQ®): Health Professional Version", PDQ Cancer Information Summaries, National Cancer Institute (US), PMID 26389282, retrieved 2018-11-26
- Guaragna MS, Soardi FC, Assumpção JG, Zambaldi L, Cardinalli IA, Yunes JA, de Mello MP, Brandalise SR, Aguiar S (August 2010). "The novel WT1 gene mutation p.H377N associated to Denys-Drash syndrome". Journal of Pediatric Hematology/Oncology. 32 (6): 486–8. doi:10.1097/MPH.0b013e3181e5e20d. PMID 20562648.
- Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH (February 1990). "Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus". Cell. 60 (3): 509–20. doi:10.1016/0092-8674(90)90601-A. PMID 2154335.
- Huff V (October 1998). "Wilms tumor genetics". American Journal of Medical Genetics. 79 (4): 260–7. doi:10.1002/(SICI)1096-8628(19981002)79:4<260::AID-AJMG6>3.0.CO;2-Q. PMID 9781905.
- Maiti S, Alam R, Amos CI, Huff V (November 2000). "Frequent association of beta-catenin and WT1 mutations in Wilms tumors". Cancer Research. 60 (22): 6288–92. PMID 11103785.
- Ruteshouser EC, Robinson SM, Huff V (June 2008). "Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors". Genes, Chromosomes & Cancer. 47 (6): 461–70. doi:10.1002/gcc.20553. PMC 4332772. PMID 18311776.
- Dome JS, Graf N, Geller JI, Fernandez CV, Mullen EA, Spreafico F, Van den Heuvel-Eibrink M, Pritchard-Jones K (September 2015). "Advances in Wilms Tumor Treatment and Biology: Progress Through International Collaboration". Journal of Clinical Oncology. 33 (27): 2999–3007. doi:10.1200/JCO.2015.62.1888. PMC 4567702. PMID 26304882.
- PDQ Pediatric Treatment Editorial Board (2002). Wilms Tumor and Other Childhood Kidney Tumors Treatment (PDQ®): Health Professional Version. PDQ Cancer Information Summaries. National Cancer Institute (US). PMID 26389282. Retrieved 2018-11-12.
- "Presentation, diagnosis, and staging of Wilms tumor".
- Szychot E, Apps J, Pritchard-Jones K (January 2014). "Wilms' tumor: biology, diagnosis and treatment". Translational Pediatrics. 3 (1): 12–24. doi:10.3978/j.issn.2224-4336.2014.01.09. PMC 4728859. PMID 26835318.
- "How is Wilms tumor staged?". www.cancer.org. Retrieved 2015-11-15.
- "Wilms Tumor - Childhood - Stages". Cancer.Net. Retrieved 2015-11-15.
- "Treatment by type and stage of Wilms tumor". www.cancer.org. Retrieved 2015-11-13.
- Stewénius Y, Jin Y, Øra I, de Kraker J, Bras J, Frigyesi A, Alumets J, Sandstedt B, Meeker AK, Gisselsson D (November 2007). "Defective chromosome segregation and telomere dysfunction in aggressive Wilms' tumors". Clinical Cancer Research. 13 (22 Pt 1): 6593–602. doi:10.1158/1078-0432.CCR-07-1081. PMID 18006759.
- Tournade MF, Com-Nougué C, de Kraker J, Ludwig R, Rey A, Burgers JM, Sandstedt B, Godzinski J, Carli M, Potter R, Zucker JM (January 2001). "Optimal duration of preoperative therapy in unilateral and nonmetastatic Wilms' tumor in children older than 6 months: results of the Ninth International Society of Pediatric Oncology Wilms' Tumor Trial and Study". Journal of Clinical Oncology. 19 (2): 488–500. doi:10.1200/jco.2001.19.2.488. PMID 11208843.
- Messahel B, Williams R, Ridolfi A, A'hern R, Warren W, Tinworth L, Hobson R, Al-Saadi R, Whyman G, Brundler MA, Kelsey A, Sebire N, Jones C, Vujanic G, Pritchard-Jones K (March 2009). "Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study". European Journal of Cancer. 45 (5): 819–26. doi:10.1016/j.ejca.2009.01.005. PMID 19231157.
- Grundy PE, Breslow NE, Li S, Perlman E, Beckwith JB, Ritchey ML, Shamberger RC, Haase GM, D'Angio GJ, Donaldson M, Coppes MJ, Malogolowkin M, Shearer P, Thomas PR, Macklis R, Tomlinson G, Huff V, Green DM (October 2005). "Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group". Journal of Clinical Oncology. 23 (29): 7312–21. doi:10.1200/JCO.2005.01.2799. PMID 16129848.
- Unless otherwise specified in boxes, then reference is: Treatment of Wilms Tumor at National Cancer Institute. Last Modified: 03/29/2012
- Spreafico F, Pritchard Jones K, Malogolowkin MH, Bergeron C, Hale J, de Kraker J, Dallorso S, Acha T, de Camargo B, Dome JS, Graf N (December 2009). "Treatment of relapsed Wilms tumors: lessons learned". Expert Review of Anticancer Therapy. 9 (12): 1807–15. doi:10.1586/era.09.159. PMID 19954292.
- Sonn G, Shortliffe LM (October 2008). "Management of Wilms tumor: current standard of care". Nature Clinical Practice. Urology. 5 (10): 551–60. doi:10.1038/ncpuro1218. PMID 18836464.
- Kalish JM, Doros L, Helman LJ, Hennekam RC, Kuiper RP, Maas SM, Maher ER, Nichols KE, Plon SE, Porter CC, Rednam S, Schultz KA, States LJ, Tomlinson GE, Zelley K, Druley TE (July 2017). "Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma". Clinical Cancer Research. 23 (13): e115–e122. doi:10.1158/1078-0432.CCR-17-0710. PMC 5538793. PMID 28674120.
- Breslow N, Olshan A, Beckwith JB, Green DM (1993). "Epidemiology of Wilms tumor". Medical and Pediatric Oncology. 21 (3): 172–81. doi:10.1002/mpo.2950210305. PMID 7680412.
- Breslow NE, Beckwith JB, Perlman EJ, Reeve AE (September 2006). "Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor". Pediatric Blood & Cancer. 47 (3): 260–7. doi:10.1002/pbc.20891. PMC 1543666. PMID 16700047.
- Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman D (July 1990). "The candidate Wilms' tumour gene is involved in genitourinary development". Nature. 346 (6280): 194–7. Bibcode:1990Natur.346..194P. doi:10.1038/346194a0. PMID 2164159.
- Nephroblastoma in childhood: current possibilities for an early radiographic diagnosis,Italian Journal of Surgery 1969
External links
| Classification | |
|---|---|
| External resources |