Poliovirus
Poliovirus, the causative agent of polio (also known as poliomyelitis), is a member virus of Enterovirus C, in the family of Picornaviridae.[1]
| Poliovirus 1 | |
|---|---|
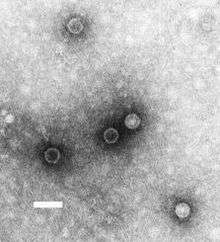 | |
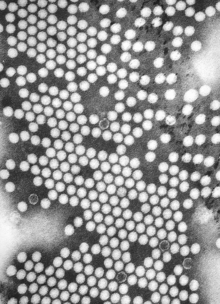
| TEM micrograph of poliovirus virions. Scale bar, 50 nm. | |
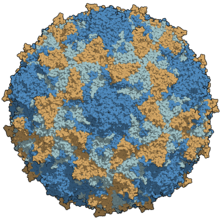 | |
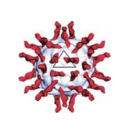
| A type 3 poliovirus capsid coloured by chains | |
| Virus classification | |
| (unranked): | Virus |
| Realm: | Riboviria |
| Phylum: | incertae sedis |
| Order: | Picornavirales |
| Family: | Picornaviridae |
| Genus: | Enterovirus |
| Species: | Enterovirus C |
| Virus: | Poliovirus 1 |
Poliovirus is composed of an RNA genome and a protein capsid. The genome is a single-stranded positive-sense RNA genome that is about 7500 nucleotides long.[2] The viral particle is about 30 nm in diameter with icosahedral symmetry. Because of its short genome and its simple composition—only RNA and a nonenveloped icosahedral protein coat that encapsulates it, poliovirus is widely regarded as the simplest significant virus.[3]
Poliovirus was first isolated in 1909 by Karl Landsteiner and Erwin Popper.[4] The structure of the virus was first elucidated using x-ray diffraction by a team at Birkbeck College led by Rosalind Franklin,[5][6] showing that the polio virus to have icosahedral symmetry.[7]
In 1981, the poliovirus genome was published by two different teams of researchers: by Vincent Racaniello and David Baltimore at MIT[8] and by Naomi Kitamura and Eckard Wimmer at Stony Brook University.[9] Poliovirus is one of the most well-characterized viruses, and has become a useful model system for understanding the biology of RNA viruses.
Replication cycle
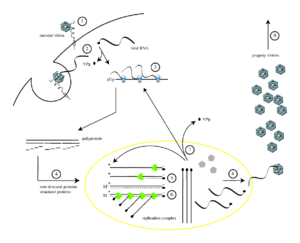
Poliovirus infects human cells by binding to an immunoglobulin-like receptor, CD155 (also known as the poliovirus receptor or PVR)[11][12] on the cell surface.[13] Interaction of poliovirus and CD155 facilitates an irreversible conformational change of the viral particle necessary for viral entry.[14][15] Attached to the host cell membrane, entry of the viral nucleic acid was thought to occur one of two ways: via the formation of a pore in the plasma membrane through which the RNA is then “injected” into the host cell cytoplasm, or that the virus is taken up by receptor-mediated endocytosis.[16] Recent experimental evidence supports the latter hypothesis and suggests that poliovirus binds to CD155 and is taken up by endocytosis. Immediately after internalization of the particle, the viral RNA is released.[17]
Poliovirus is a positive-stranded RNA virus. Thus, the genome enclosed within the viral particle can be used as messenger RNA and immediately translated by the host cell. On entry, the virus hijacks the cell's translation machinery, causing inhibition of cellular protein synthesis in favor of virus-specific protein production.[18] Unlike the host cell's mRNAs, the 5' end of poliovirus RNA is extremely long—over 700 nucleotides—and highly structured. This region of the viral genome is called internal ribosome entry site (IRES), and it directs translation of the viral RNA. Genetic mutations in this region prevent viral protein production.[19][20][21] The first IRES to be discovered was found in poliovirus RNA.[22]
Poliovirus mRNA is translated as one long polypeptide. This polypeptide is then autocleaved by internal proteases into about 10 individual viral proteins. Not all cleavages occur with the same efficiency. Therefore, the amounts of proteins produced by the polypeptide cleavage vary: for example, smaller amounts of 3Dpol are produced than those of capsid proteins, VP1–4.[23][24] These individual viral proteins are:[3][25]
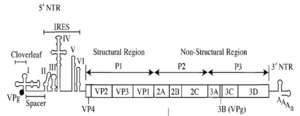
- 3Dpol, an RNA dependent RNA polymerase whose function is to make multiple copies of the viral RNA genome
- 2Apro and 3Cpro/3CDpro, proteases which cleave the viral polypeptide
- VPg (3B), a small protein that binds viral RNA and is necessary for synthesis of viral positive and negative strand RNA
- 2BC, 2B, 2C (an ATPase)[26], 3AB, 3A, 3B proteins which comprise the protein complex needed for virus replication.
- VP0, which is further cleaved into VP2 and VP4, VP1 and VP3, proteins of the viral capsid
After translation, transcription and genome replication which involve a single process (synthesis of (+) RNA) is realized. For the infecting (+) RNA to be replicated, multiple copies of (−) RNA must be transcribed and then used as templates for (+) RNA synthesis. Replicative intermediates (RIs) which are an association of RNA molecules consisting of a template RNA and several growing RNAs of varying length, are seen in both the replication complexes for (−) RNAs and (+) RNAs. The primer for both (+) and (−) strand synthesis is the small protein VPg, which is uridylylated at the hydroxyl group of a tyrosine residue by the poliovirus RNA polymerase at a cis-acting replication element located in a stem-loop in the virus genome. Some of the (+) RNA molecules are used as templates for further (−) RNA synthesis, some function as mRNA, and some are destined to be the genomes of progeny virions.[23]
In the assembly of new virus particles (i.e. the packaging of progeny genome into a procapsid which can survive outside the host cell), including, respectively:[27]
- Five copies each of VP0, VP3, and VP1 which its N termini and VP4 form interior surface of capsid, assemble into a ‘pentamer’ and 12 pentamers form a procapsid. (The outer surface of capsid is consisting of VP1, VP2, VP3; C termini of VP1 and VP3 form the canyons which around each of the vertices; around this time, the 60 copies of VP0 are cleaved into VP4 and VP2.)
- Each procapsid acquires a copy of the virus genome, with VPg still attached at the 5' end.
Fully assembled poliovirus leaves the confines of its host cell by lysis[28] 4 to 6 hours following initiation of infection in cultured mammalian cells.[29] The mechanism of viral release from the cell is unclear,[2] but each dying cell can release up to 10,000 polio virions.[29]
Drake demonstrated that poliovirus is able to undergo multiplicity reactivation.[30] That is, when polioviruses were irradiated with UV light and allowed to undergo multiple infections of host cells, viable progeny could be formed even at UV doses that inactivated the virus in single infections.
Origin and serotypes
Poliovirus is structurally similar to other human enteroviruses (coxsackieviruses, echoviruses, and rhinoviruses), which also use immunoglobulin-like molecules to recognize and enter host cells.[12] Phylogenetic analysis of the RNA and protein sequences of poliovirus suggests that it may have evolved from a C-cluster Coxsackie A virus ancestor, that arose through a mutation within the capsid.[31] The distinct speciation of poliovirus probably occurred as a result of change in cellular receptor specificity from intercellular adhesion molecule-1 (ICAM-1), used by C-cluster Coxsackie A viruses, to CD155; leading to a change in pathogenicity, and allowing the virus to infect nervous tissue.
The mutation rate in the virus is relatively high even for an RNA virus with a synonymous substitution rate of 1.0 x 10−2 substitutions/site/year and non synonymous substitution rate of 3.0 x 10−4 substitutions/site/year.[32] Base distribution within the genome is not random with adenosine being less common than expected at the 5' end and higher at the 3' end.[33] Codon use is not random with codons ending in adenosine being favoured and those ending in cytosine or guanine being avoided. Codon use differs between the three genotypes and appears to be driven by mutation rather than selection.[34]
The three serotypes of poliovirus, PV1, PV2, and PV3, each have a slightly different capsid protein. Capsid proteins define cellular receptor specificity and virus antigenicity. PV1 is the most common form encountered in nature, but all three forms are extremely infectious.[4] As of November 2015, wild PV1 is highly localized to regions in Pakistan and Afghanistan. Wild PV2 was declared eradicated in September 2015 after last being detected in October 1999 in Uttar Pradesh, India.[35] As of November 2015, wild PV3 has not been seen since its 2012 detection in parts of Nigeria and Pakistan.[36]
Specific strains of each serotype are used to prepare vaccines against polio. Inactive polio vaccine is prepared by formalin inactivation of three wild, virulent reference strains, Mahoney or Brunenders (PV1), MEF-1/Lansing (PV2), and Saukett/Leon (PV3). Oral polio vaccine contains live attenuated (weakened) strains of the three serotypes of poliovirus. Passaging the virus strains in monkey kidney epithelial cells introduces mutations in the viral IRES, and hinders (or attenuates) the ability of the virus to infect nervous tissue.[29]
Polioviruses were formerly classified as a distinct species belonging to the genus Enterovirus in the family Picornaviridae. In 2008, the Poliovirus species was eliminated and the three serotypes were assigned to the species Human enterovirus C (later renamed Enterovirus C), in the genus Enterovirus in the family Picornaviridae. The type species of the genus Enterovirus was changed from Poliovirus to (Human) Enterovirus C.[37]
Pathogenesis
The primary determinant of infection for any virus is its ability to enter a cell and produce additional infectious particles. The presence of CD155 is thought to define the animals and tissues that can be infected by poliovirus. CD155 is found (outside of laboratories) only on the cells of humans, higher primates, and Old World monkeys. Poliovirus is, however, strictly a human pathogen, and does not naturally infect any other species (although chimpanzees and Old World monkeys can be experimentally infected).[38]
The CD155 gene appears to have been subject to positive selection.[39] The protein has several domains of which domain D1 contains the polio virus binding site. Within this domain, 37 amino acids are responsible for binding the virus.
Poliovirus is an enterovirus. Infection occurs via the fecal–oral route, meaning that one ingests the virus and viral replication occurs in the alimentary tract.[40] Virus is shed in the feces of infected individuals. In 95% of cases only a primary, transient presence of viremia (virus in the bloodstream) occurs, and the poliovirus infection is asymptomatic. In about 5% of cases, the virus spreads and replicates in other sites such as brown fat, reticuloendothelial tissue, and muscle. The sustained viral replication causes secondary viremia and leads to the development of minor symptoms such as fever, headache, and sore throat.[41] Paralytic poliomyelitis occurs in less than 1% of poliovirus infections. Paralytic disease occurs when the virus enters the central nervous system (CNS) and replicates in motor neurons within the spinal cord, brain stem, or motor cortex, resulting in the selective destruction of motor neurons leading to temporary or permanent paralysis. In rare cases, paralytic poliomyelitis leads to respiratory arrest and death. In cases of paralytic disease, muscle pain and spasms are frequently observed prior to onset of weakness and paralysis. Paralysis typically persists from days to weeks prior to recovery.[42]
In many respects, the neurological phase of infection is thought to be an accidental diversion of the normal gastrointestinal infection.[16] The mechanisms by which poliovirus enters the CNS are poorly understood. Three nonmutually exclusive hypotheses have been suggested to explain its entry. All theories require primary viremia. The first hypothesis predicts that virions pass directly from the blood into the central nervous system by crossing the blood–brain barrier independent of CD155.[43] A second hypothesis suggests that the virions are transported from peripheral tissues that have been bathed in the viremic blood, for example muscle tissue, to the spinal cord through nerve pathways via retrograde axonal transport.[44][45][46] A third hypothesis is that the virus is imported into the CNS via infected monocytes or macrophages.[10]
Poliomyelitis is a disease of the central nervous system. However, CD155 is believed to be present on the surface of most or all human cells. Therefore, receptor expression does not explain why poliovirus preferentially infects certain tissues. This suggests that tissue tropism is determined after cellular infection. Recent work has suggested that the type I interferon response (specifically that of interferon alpha and beta) is an important factor that defines which types of cells support poliovirus replication.[47] In mice expressing CD155 (through genetic engineering) but lacking the type I interferon receptor, poliovirus not only replicates in an expanded repertoire of tissue types, but these mice are also able to be infected orally with the virus.[48]
Immune system avoidance
Poliovirus uses two key mechanisms to evade the immune system. First, it is capable of surviving the highly acidic conditions of the stomach, allowing the virus to infect the host and spread throughout the body via the lymphatic system.[3] Second, because it can replicate very quickly, the virus overwhelms the host organs before an immune response can be mounted.[49] If detail is given at the attachment phase; poliovirus with canyons on the virion surface have virus attachment sites located in pockets at the canyon bases. The canyons are too narrow for access by antibodies, so the virus attachment sites are protected from the host’s immune surveillance, while the remainder of the virion surface can mutate to avoid the host’s immune response.[50]
Individuals who are exposed to poliovirus, either through infection or by immunization with polio vaccine, develop immunity. In immune individuals, antibodies against poliovirus are present in the tonsils and gastrointestinal tract (specifically IgA antibodies) and are able to block poliovirus replication; IgG and IgM antibodies against poliovirus can prevent the spread of the virus to motor neurons of the central nervous system.[29] Infection with one serotype of poliovirus does not provide immunity against the other serotypes, however second attacks within the same individual are extremely rare.
PVR transgenic mouse
Although humans are the only known natural hosts of poliovirus, monkeys can be experimentally infected and they have long been used to study poliovirus. In 1990–91, a small animal model of poliomyelitis was developed by two laboratories. Mice were engineered to express a human receptor to poliovirus (hPVR).[51][52]
Unlike normal mice, transgenic poliovirus receptor (TgPVR) mice are susceptible to poliovirus injected intravenously or intramuscularly, and when injected directly into the spinal cord or the brain.[53] Upon infection, TgPVR mice show signs of paralysis that resemble those of poliomyelitis in humans and monkeys, and the central nervous systems of paralyzed mice are histocytochemically similar to those of humans and monkeys. This mouse model of human poliovirus infection has proven to be an invaluable tool in understanding poliovirus biology and pathogenicity.[54]
Three distinct types of TgPVR mice have been well studied:[55]
- In TgPVR1 mice, the transgene encoding the human PVR was incorporated into mouse chromosome 4. These mice express the highest levels of the transgene and the highest sensitivity to poliovirus. TgPVR1 mice are susceptible to poliovirus through the intraspinal, intracerebral, intramuscular, and intravenous pathways, but not through the oral route.
- TgPVR21 mice have incorporated the human PVR at chromosome 13. These mice are less susceptible to poliovirus infection through the intracerebral route, possibly because they express decreased levels of hPVR. TgPVR21 mice have been shown to be susceptible to poliovirus infection through intranasal inoculation, and may be useful as a mucosal infection model.[56]
- In TgPVR5 mice, the human transgene is located on chromosome 12. These mice exhibit the lowest levels of hPVR expression and are the least susceptible to poliovirus infection.
Recently, a fourth TgPVR mouse model was developed. These "cPVR" mice carry hPVR cDNA, driven by a β-actin promoter, and have proven susceptible to poliovirus through intracerebral, intramuscular, and intranasal routes. In addition, these mice are capable of developing the bulbar form of polio after intranasal inoculation.[56]
The development of the TgPVR mouse has had a profound effect on oral poliovirus vaccine (OPV) production. Previously, monitoring the safety of OPV had to be performed using monkeys, because only primates are susceptible to the virus. In 1999, the World Health Organization approved the use of the TgPVR mouse as an alternative method of assessing the effectiveness of the vaccine against poliovirus type-3. In 2000, the mouse model was approved for tests of vaccines against type-1 and type-2 poliovirus.[57]
Cloning and synthesis
In 1981, Racaniello and Baltimore used recombinant DNA technology to generate the first infectious clone of an animal RNA virus, poliovirus. DNA encoding the RNA genome of poliovirus was introduced into cultured mammalian cells and infectious poliovirus was produced.[58] Creation of the infectious clone propelled understanding of poliovirus biology, and has become a standard technology used to study many other viruses.
In 2002, Eckard Wimmer's group at Stony Brook University succeeded in synthesizing poliovirus from its chemical code, producing the world's first synthetic virus.[59] Scientists first converted poliovirus's published RNA sequence, 7741 bases long, into a DNA sequence, as DNA was easier to synthesize. Short fragments of this DNA sequence were obtained by mail-order, and assembled. The complete viral genome was then assembled by a gene synthesis company. This whole painstaking process took two years. Nineteen markers were incorporated into the synthesized DNA, so that it could be distinguished from natural poliovirus. Enzymes were used to convert the DNA back into RNA, its natural state. Other enzymes were then used to translate the RNA into a polypeptide, producing functional viral particle. The newly minted synthetic virus was injected into PVR transgenic mice, to determine if the synthetic version was able to cause disease. The synthetic virus was able to replicate, infect, and cause paralysis or death in mice. However, the synthetic version was between 1/1,000th and 1/10,000th as lethal as the original virus.[60]
Modification for therapies
A modification of the poliovirus, called PVSRIPO, was tested in early clinical trials as a possible treatment for cancer.[61]
References
- Ryan KJ, Ray CG, eds. (2004). Sherris Medical Microbiology (4th ed.). McGraw Hill. ISBN 978-0-8385-8529-0.
- Hogle J (2002). "Poliovirus cell entry: common structural themes in viral cell entry pathways". Annu Rev Microbiol. 56: 677–702. doi:10.1146/annurev.micro.56.012302.160757. PMC 1500891. PMID 12142481.
- Goodsell DS (1998). The machinery of life. New York: Copernicus. ISBN 978-0-387-98273-1.
- Paul JR (1971). A History of Poliomyelitis. (Yale studies in the history of science and medicine). New Haven, Conn: Yale University Press. ISBN 978-0-300-01324-5.
- "Behind the picture: Rosalind Franklin and the polio model". Medical Research Council. 2019-03-14. Retrieved 4 September 2019.
- Maddox, Brenda (2003). Rosalind Franklin: The Dark Lady of DNA. London: Harper Collins. p. 296. ISBN 0-00-655211-0.
- Brown, Andrew (2007). J.D. Bernal: The Sage of Science. New York: Oxford University Press. pp. 359–361. ISBN 978-0-19-920565-3.
- Racaniello and Baltimore; Baltimore, D (1981). "Molecular cloning of poliovirus cDNA and determination of the complete nucleotide sequence of the viral genome". Proc. Natl. Acad. Sci. U.S.A. 78 (8): 4887–91. Bibcode:1981PNAS...78.4887R. doi:10.1073/pnas.78.8.4887. PMC 320284. PMID 6272282.
- Kitamura N, Semler B, Rothberg P, et al. (1981). "Primary structure, gene organization and polypeptide expression of poliovirus RNA". Nature. 291 (5816): 547–53. Bibcode:1981Natur.291..547K. doi:10.1038/291547a0. PMID 6264310.
- De Jesus NH (2007). "Epidemics to eradication: the modern history of poliomyelitis". Virol. J. 4 (1): 70. doi:10.1186/1743-422X-4-70. PMC 1947962. PMID 17623069.
- Mendelsohn Cl; Wimmer E; Racaniello VR (1989). "Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobin superfamily". Cell. 56 (5): 855–865. doi:10.1016/0092-8674(89)90690-9. PMID 2538245.
- He Y, Mueller S, Chipman P, et al. (2003). "Complexes of poliovirus serotypes with their common cellular receptor, CD155". J Virol. 77 (8): 4827–35. doi:10.1128/JVI.77.8.4827-4835.2003. PMC 152153. PMID 12663789.
- Dunnebacke TH, Levinthal JD, Williams RC (1 October 1969). "Entry and release of poliovirus as observed by electron microscopy of cultured cells". J. Virol. 4 (4): 505–13. PMC 375900. PMID 4309884.
- Kaplan G, Freistadt MS, Racaniello VR (1 October 1990). "Neutralization of poliovirus by cell receptors expressed in insect cells". J. Virol. 64 (10): 4697–702. PMC 247955. PMID 2168959.
- Gómez Yafal A, Kaplan G, Racaniello VR, Hogle JM (November 1993). "Characterization of poliovirus conformational alteration mediated by soluble cell receptors". Virology. 197 (1): 501–5. doi:10.1006/viro.1993.1621. PMID 8212594.
- Mueller S, Wimmer E, Cello J (2005). "Poliovirus and poliomyelitis: a tale of guts, brains, and an accidental event". Virus Res. 111 (2): 175–93. doi:10.1016/j.virusres.2005.04.008. PMID 15885840.
- Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM (2007). "Imaging poliovirus entry in live cells". PLOS Biology. 5 (7): e183. doi:10.1371/journal.pbio.0050183. PMC 1914398. PMID 17622193.
- Attardi G, Smith J (1962). "Virus specific protein and a ribo-nucleic acid associated with ribosomes in poliovirus infected HeLa cells". Cold Spring Harb. Symp. Quant. Biol. 27: 271–92. doi:10.1101/SQB.1962.027.001.026. PMID 13965389.
- Chen CY, Sarnow P (1995). "Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs". Science. 268 (5209): 415–7. Bibcode:1995Sci...268..415C. doi:10.1126/science.7536344. PMID 7536344.
- Pelletier J, Sonenberg N (1988). "Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA". Nature. 334 (6180): 320–5. Bibcode:1988Natur.334..320P. doi:10.1038/334320a0. PMID 2839775.
- Jang SK, Kräusslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E (1 August 1988). "A segment of the 5' nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation". J. Virol. 62 (8): 2636–43. PMC 253694. PMID 2839690.
- John Carter; Venetia A. Saunders (2007). Virology: Principles and Applications. John Wiley & Sons. p. 4. ISBN 978-0-470-02386-0.
- John Carter; Venetia A. Saunders (2007). Virology: Principles and Applications. John Wiley & Sons. p. 165. ISBN 978-0-470-02386-0.
- Harper, David R. (2012). Viruses: Biology, Applications, and Control. The United States of America: Garland Science. p. 57. ISBN 978-0-8153-4150-5.
- Charles Chan and Roberto Neisa. "Poliomyelitis". Archived February 22, 2007, at the Wayback Machine Brown University.
- John Carter; Venetia A. Saunders (2007). Virology: Principles and Applications. John Wiley & Sons. p. 164. ISBN 978-0-470-02386-0.
- John Carter; Venetia A. Saunders (2007). Virology: Principles and Applications. John Wiley & Sons. pp. 161, 165. ISBN 978-0-470-02386-0.
- John Carter; Venetia A. Saunders (2007). Virology: Principles and Applications. John Wiley & Sons. p. 166. ISBN 978-0-470-02386-0.
- Kew O, Sutter R, de Gourville E, Dowdle W, Pallansch M (2005). "Vaccine-derived polioviruses and the endgame strategy for global polio eradication". Annu Rev Microbiol. 59: 587–635. doi:10.1146/annurev.micro.58.030603.123625. PMID 16153180.
- DRAKE JW (August 1958). "Interference and multiplicity reactivation in polioviruses". Virology. 6 (1): 244–64. doi:10.1016/0042-6822(58)90073-4. PMID 13581529.
- Jiang P, Faase JA, Toyoda H, et al. (2007). "Evidence for emergence of diverse polioviruses from C-cluster Coxsackie A viruses and implications for global poliovirus eradication". Proc. Natl. Acad. Sci. U.S.A. 104 (22): 9457–62. Bibcode:2007PNAS..104.9457J. doi:10.1073/pnas.0700451104. PMC 1874223. PMID 17517601.
- Jorba J, Campagnoli R, De L, Kew O (2008). "Calibration of multiple poliovirus molecular clocks covering an extended evolutionary range". J. Virol. 82 (9): 4429–40. doi:10.1128/JVI.02354-07. PMC 2293050. PMID 18287242.
- Rothberg PG, Wimmer E (1981). "Mononucleotide and dinucleotide frequencies, and codon usage in poliovirion RNA". Nucleic Acids Res. 9 (23): 6221–9. doi:10.1093/nar/9.23.6221. PMC 327599. PMID 6275352.
- Zhang J, Wang M, Liu WQ, et al. (2011). "Analysis of codon usage and nucleotide composition bias in polioviruses". Virol. J. 8: 146. doi:10.1186/1743-422X-8-146. PMC 3079669. PMID 21450075.
- "Global eradication of wild poliovirus type 2 declared". Global Polio Eradication Initiative. 2015-09-20. Retrieved 2015-09-30.
- "Possible Eradication of Wild Poliovirus Type 3 — Worldwide, 2012". CDC. 2014-11-14. Retrieved 2014-11-14.
- Carstens, E. B.; Ball, L. A. (July 2009). "Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2008)". Archives of Virology. 154 (7): 1181–8. doi:10.1007/s00705-009-0400-2. ISSN 1432-8798. PMID 19495937.
- Mueller S, Wimmer E (2003). "Recruitment of nectin-3 to cell-cell junctions through trans-heterophilic interaction with CD155, a vitronectin and poliovirus receptor that localizes to alpha(v)beta3 integrin-containing membrane microdomains". J Biol Chem. 278 (33): 31251–60. doi:10.1074/jbc.M304166200. PMID 12759359.
- Suzuki Y (2006). "Ancient positive selection on CD155 as a possible cause for susceptibility to poliovirus infection in simians". Gene. 373: 16–22. doi:10.1016/j.gene.2005.12.016. PMID 16500041.
- Bodian D, Horstmann DH (1969). Polioviruse. Philadelphia, Penn: Lippincott. pp. 430–73.
- Sabin A (1956). "Pathogenesis of poliomyelitis; reappraisal in the light of new data". Science. 123 (3209): 1151–7. Bibcode:1956Sci...123.1151S. doi:10.1126/science.123.3209.1151. PMID 13337331.
- Acute Poliomyelitis at eMedicine
Pediatric Poliomyelitis at eMedicine - Yang W, Terasaki T, Shiroki K, et al. (1997). "Efficient delivery of circulating poliovirus to the central nervous system independently of poliovirus receptor". Virology. 229 (2): 421–8. doi:10.1006/viro.1997.8450. PMID 9126254.
- Ohka S. Yang WX; Terada E; Iwasaki K; Nomot A (1998). "Retrograde transport of intact poliovirus through the axon via the first transport system". Virology. 250 (1): 67–75. doi:10.1006/viro.1998.9360. PMID 9770421.
- Ren R, Racaniello V (1992). "Poliovirus spreads from muscle to the central nervous system by neural pathways". J Infect Dis. 166 (4): 747–52. doi:10.1093/infdis/166.4.747. PMID 1326581.
- Lancaster KZ, Pfeiffer JK (2010). Gale, Michael (ed.). "Limited trafficking of a neurotropic virus through inefficient retrograde axonal transport and the type I interferon response". PLoS Pathog. 6 (3): e1000791. doi:10.1371/journal.ppat.1000791. PMC 2832671. PMID 20221252.
- Ida-Hosonuma M, Iwasaki T, Yoshikawa T, et al. (April 2005). "The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus". J. Virol. 79 (7): 4460–9. doi:10.1128/JVI.79.7.4460-4469.2005. PMC 1061561. PMID 15767446.
- Ohka S, Igarashi H, Sakai M, Koike S, Nochi T, Kiyono A, Nomoto A (2007). "Establishment of a poliovirus oral infection system in human poliovirus receptor-expressing transgenic mice that are deficient in alpha/beta interferon receptor". J. Virol. 81 (15): 7902–12. doi:10.1128/JVI.02675-06. PMC 1951287. PMID 17507470.
- Racaniello V (2006). "One hundred years of poliovirus pathogenesis". Virology. 344 (1): 9–16. doi:10.1016/j.virol.2005.09.015. PMID 16364730.
- Carter, John B. Saunders, Venetia A. (2007). Virology: Principles and Applications. Liverpool John Moores University, UK: John Wiley & Sons. p. 162. ISBN 978-0-470-02386-0.
- Ren RB, Costantini F, Gorgacz EJ, Lee JJ, Racaniello VR (1990). "Transgenic mice expressing a human poliovirus receptor: a new model for poliomyelitis". Cell. 63 (2): 353–62. doi:10.1016/0092-8674(90)90168-E. PMID 2170026.
- Koike S, Taya C, Kurata T, et al. (1991). "Transgenic mice susceptible to poliovirus". Proc. Natl. Acad. Sci. U.S.A. 88 (3): 951–5. Bibcode:1991PNAS...88..951K. doi:10.1073/pnas.88.3.951. PMC 50932. PMID 1846972.
- Horie H, Koike S, Kurata T, et al. (1 February 1994). "Transgenic mice carrying the human poliovirus receptor: new animal models for study of poliovirus neurovirulence". J. Virol. 68 (2): 681–8. PMC 236503. PMID 8289371.
- Ohka S, Nomoto A (2001). "Recent insights into poliovirus pathogenesis". Trends Microbiol. 9 (10): 501–6. doi:10.1016/S0966-842X(01)02200-4. PMID 11597452.
- Koike S, Taya C, Aoki J, et al. (1994). "Characterization of three different transgenic mouse lines that carry human poliovirus receptor gene—influence of the transgene expression on pathogenesis". Arch. Virol. 139 (3–4): 351–63. doi:10.1007/BF01310797. PMID 7832641.
- Nagata N, Iwasaki T, Ami Y, Sato Y, Hatano I, Harashima A, et al. (March 2004). "A poliomyelitis model through mucosal infection in transgenic mice bearing human poliovirus receptor, TgPVR21". Virology. 321 (1): 87–100. doi:10.1016/j.virol.2003.12.008. PMID 15033568.
- Dragunsky E, Nomura T, Karpinski K, et al. (2003). "Transgenic mice as an alternative to monkeys for neurovirulence testing of live oral poliovirus vaccine: validation by a WHO collaborative study". Bull. World Health Organ. 81 (4): 251–60. doi:10.1590/S0042-96862003000400006 (inactive 2019-12-11). PMC 2572431. PMID 12764491.
- Racaniello V, Baltimore D (1981). "Cloned poliovirus complemenatry DNA is infectious in mammalian cells". Science. 214 (453): 916–9. Bibcode:1981Sci...214..916R. doi:10.1126/science.6272391. PMID 6272391.
- Cello J, Paul AV, Wimmer E (2002). "Chemical synthesis of poliovirus cDNA: generation of infectious virus in the absence of natural template". Science. 297 (5583): 1016–8. Bibcode:2002Sci...297.1016C. doi:10.1126/science.1072266. PMID 12114528.
- Couzin J (2002). "Virology. Active poliovirus baked from scratch". Science. 297 (5579): 174–5. doi:10.1126/science.297.5579.174b. PMID 12114601.
- Brown MC, Dobrikova EY, Dobrikov MI, Walton RW, Gemberling SL, Nair SK, Desjardins A, Sampson JH, Friedman HS, Friedman AH, Tyler DS, Bigner DD, Gromeier M (November 2014). "Oncolytic polio virotherapy of cancer". Cancer. 120 (21): 3277–86. doi:10.1002/cncr.28862. PMC 4205207. PMID 24939611.
External links
- ICTVdb virus classification 2006
- Home of Picornaviruses (latest updates of species, serotypes, & proposed changes)
- Goodsell, David. "Poliovirus and Rhinovirus". August 2001 Molecule of the Month.
- 3D macromolecular structures of the Poliovirus archived in the EM Data Bank(EMDB)
- "Human poliovirus 1". NCBI Taxonomy Browser. 12080.
- "Human poliovirus 3". NCBI Taxonomy Browser. 12086.