Floating–Harbor syndrome
Floating–Harbor syndrome, also known as Pelletier–Leisti syndrome, is a rare disease with fewer than 50 cases described in the literature.[1] It is usually diagnosed in early childhood and is characterized by the triad of proportionate short stature with delayed bone age, characteristic facial appearance, and delayed speech development.[2][3] Although its cause is unknown, it is thought to result from genetic mutation, and diagnosis is established by the presence of a heterozygous SRCAP mutation in those with clinical findings of FHS.[4]
| Floating–Harbor syndrome | |
|---|---|
| Other names | Short stature with delayed bone age, expressive language delay, a triangular face with a prominent nose and deep-set eyes, FHS |
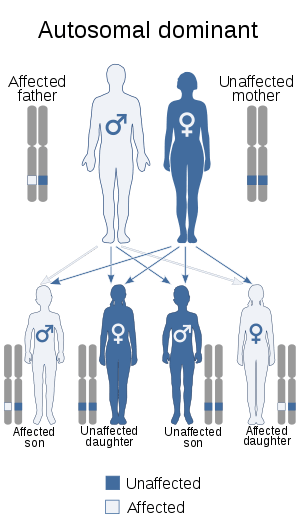 | |
| This condition is inherited via autosomal dominant manner | |
| Specialty | Medical genetics |
Signs and symptoms
Below are the common clinical features of those diagnosed with Floating–Harbor syndrome. Patients will show varying degrees of some or all FHS symptoms. Facial abnormalities are the most defining aspects of those diagnosed with this disease.
Cardinal facial features
Voice quality and language
- Dysarthria and verbal dyspraxia with phoneme imprecision
- Hypernasality
- High-pitched voices
- Severe receptive and expressive language impairment across all domains of function[4]
Bodily features
- Significant delay in bone age (-2 SD or greater) with normalization between 6–12 years old
- Skeletal anomalies: brachydactyly, broad fingertips or clubbing, clinodactyly, short thumbs, prominent joints, clavicle abnormalities
- Short adult stature: for females, maximum height was at the 20th percentile; for males, the maximum height was at the 25th percentile, though male height varied more widely[5]
The differential diagnosis of broad thumbs includes Rubinstein-taybi syndrome, where they are a cardinal feature. FHS is also in the differential, which logically agrees with the thought that the disease is a result of a mutation in SRCAP, as this gene interacts with CBP.[4]
Behavior
- Tantrums during infancy
- Attention deficit-hyperactivity disorder (ADHD) during school years: impulsivity, inattention, restlessness
- Unpredictable, aggressive outbursts
- Autistic spectrum disorder (one case)
- Asperger syndrome (one case)
- Obsessive compulsive disorder (two cases)[1][4]
Other observations
- Intellectual disability: in all cases each individual showed a varying degree of intellectual impairment and learning disability, ranging from borderline normal to moderate intellectual disability
- Early entry into puberty is shown in some girls, leading to menorrhagia and irregular periods[1]
- Dental problems (caries, malocclusion, dysplastic, small teeth)[1]
- Visual impairment[4]
Mechanism
The cause of this condition is unknown but evidence of familial inheritance and sporadic genetic mutation has been linked to cases of FHS. Two possibly familial cases have been reported—one in a mother and son,[6] and the other in a mother and daughter.[7][8] This suggests an autosomal dominant inheritance but additional cases need to be investigated to establish this. Another report has suggested that the inheritance may be autosomal recessive.[9] In all of these cases, however, the mothers and children were not similarly affected, suggesting a variable clinical expression of the syndrome.[3]
In a study published by the American Journal of Human Genetics in 2012, exome sequencing was used to investigate a group of unrelated individuals with classic features of FHS and identified heterozygous mutations in SRCAP as causative of this disorder.[10] Each reported mutation was truncating (nonsense or frameshift) and occurred between codons 2,407 and 2,517 in exon 34, resulting in the loss of three C-terminal AT-hook motifs. SRCAP encodes a SNF2-related chromatin-remodeling ATPase that is a coactivator for CREB-binding protein (or CBP), which is the major cause of Rubinstein–Taybi syndrome. This disrupted interaction between the proteins most likely explains the clinical overlap between FHS and RTS.[5]
- SRCAP has been shown to transduce signals of nuclear (steroid) hormone receptors and Notch pathways, showing that it plays diverse roles in gene expression.[10]
- SRCAP contains several functional domains (SNF2 like ATPase, an N-terminal HSA domain, and three C-terminal AT-hook DNA-binding motifs).
- The CBP interaction domain of SRCAP is located centrally.[10]
Thus, the mechanism of disease in FHS is suspected to be dominant-negative (or antimorphic) due to the mutation in the final exon that results in the loss of the major transactivation function of SRCAP (or loss of one or more critical domains). All of the patients that carried the mutation also had obvious physical symptoms (i.e., prominent nose, delayed bone age, and short stature). Those who tested negative for the mutation often had dysmorphic facial features distinct from classical FHS, as well as a formal diagnosis of autism.
Drawbacks to findings
From the same group, three individuals whose phenotype most closely resembled FHS carried no mutation on the SRCAP gene.[5] Though these findings are a large step in determining the underlying cause of FHS and widely accepted, others would claim molecular diagnosis is not always successful and the mutation is not a mandatory feature for the diagnosis of FHS.[11]
Diagnosis
Until recently, doctors have diagnosed patients with FHS based on clinical observations and how well they fit the disease description, usually occurring in early childhood. Molecular genetic testing is also used now to test for genetic mutations. By performing a sequence analysis test of select exons, mutations can be detected in exon 34 of the SRCAP gene. This mutation has been observed in 19 patients to date.[4] In most cases, if the patient shows classic facial features of FHS, the molecular testing will show a mutation on the SRCAP gene.[5]
Differential diagnosis
FHS shares some common features with Rubinstein–Taybi (due to overlapping effects of mutations on SRCAP), however cranial and hand anomalies are distinctive: broad thumbs, narrow palate, and microcephaly are absent in Floating-Harbor Syndrome.[12][13] One child in the UK has a diagnosis of microcephaly alongside Floating–Harbor syndrome.[14]
Management
There are no cures for FHS. Close monitoring of growth in the first few years is essential, as well as annual general health screening and tests listed below. An FHS diagnosis will affect the individual and those there to support them.
Managing symptoms and features of FHS involves maintaining a close watch on the patient's physical as well as mental health. This would include:
- Sequencing of SRCAP exons 31–34 in all suspected cases
- Complete assessments of auditory and visual systems
- Renal and urinary tract ultrasound
- Orthopedic assessment of hip dysplasia and clavicle abnormalities[4]
- Neurologic assessment if there is a suspicion of seizures
- Dental hygiene to prevent cavities and to monitor for malocclusion
- Evaluation for growth hormone deficiency at baseline, to be repeated if loss of growth velocity occurs
- Monitoring of bone age and pubertal timing in case of precocious puberty
- Psychoeducational assessments corrected for deficiencies in expressive language and sensory issues
- Monitoring of behavioral disturbances and provision of early intervention
- Counseling for families regarding recurrence risk and the offspring of individuals with FHS[5]
Special education programs and vocational training to address developmental disabilities are highly recommended, as well as communication rehabilitation with sign language or alternative means of communication. Behavior management strategies could also include referrals to behavior specialists or psychologists for help. For those concerned, genetic counseling can be sought for issues related to testing of at-risk relatives.
History
The first identified instances occurred in 1973 at the Boston Floating Hospital,[15] and in 1975 at Harbor General Hospital in Torrance, California.[16] The name Floating–Harbor syndrome was coined by Robinson et al. in 1988.[12] Since then approximately 40 more cases have been described.
The first case recorded was that of a 5-year-old boy. In a 32-year follow up done in 2006,[17] the patient was in good overall health, had never been hospitalized, and had been employed for the past 15 years. His mother stated he had a very good memory, was gregarious, had a temper, and at times was stubborn. Changes in the patient's facial configuration and body could be attributed to age and familial history (i.e., the patient shows signs of arthritis and hypertension). Still present were the low hairline, broad tip of the nose, short nasal labial distance, depressed columella, thin lips, and posteriorly positioned ears, as well as short stature and mild to moderate retardation.
Recent research
In a study published in 2012 in the Journal of Pediatric Endocrinology, a group of scientists reported the long-term effects of a patient diagnosed with FHS undergoing growth hormone therapy from the age of 3.5 years to 9 years old. While the GH seemed to work initially, the patient's growth after the first couple years slowed significantly and the patient reached a stable height far below the target or standard height.[18] The results on GH therapy remain inconclusive.
Recent research mostly centers around the search and confirmation of the gene responsible for FHS. As discussed in the mechanisms section, though the mutation of SRCAP is a widely accepted indicator of a patient diagnosed with FHS, it is not the cause in every case.[5][11][19]
References
- White SM, Morgan A, Da Costa A, Lacombe D, Knight SJ, Houlston R, et al. (April 2010). "The phenotype of Floating-Harbor syndrome in 10 patients". American Journal of Medical Genetics. Part A. 152A (4): 821–9. doi:10.1002/ajmg.a.33294. PMID 20358590. (subscription required)
- Bastaki L, El-Nabi MM, Azab AS, Gouda SA, Al-Wadaani AM, Naguib KK (2007). "Floating-Harbor syndrome in a Kuwaiti patient: a case report and literature review". Eastern Mediterranean Health Journal. 13 (4): 975–9. PMID 17955782.
- Arpin S, Afenjar A, Dubern B, Toutain A, Cabrol S, Héron D (January 2012). "Floating-Harbor Syndrome: report on a case in a mother and daughter, further evidence of autosomal dominant inheritance". Clinical Dysmorphology. 21 (1): 11–4. doi:10.1097/mcd.0b013e32834af5a7. PMID 21955542.
- Nowaczyk MJ (1993). "Floating-Harbor Syndrome". University of Washington. Retrieved 1 April 2014.
- Nikkel SM, Dauber A, de Munnik S, Connolly M, Hood RL, Caluseriu O, et al. (April 2013). "The phenotype of Floating-Harbor syndrome: clinical characterization of 52 individuals with mutations in exon 34 of SRCAP". Orphanet Journal of Rare Diseases. 8: 63. doi:10.1186/1750-1172-8-63. PMC 3659005. PMID 23621943.
- Peñaloza JM, García-Cruz D, Dávalos IP, Dávalos NO, García-Cruz MO, Pérez-Rulfo D, Sánchez-Corona J (2003). "A variant example of familial Floating-Harbor syndrome?". Genetic Counseling. 14 (1): 31–7. PMID 12725587.
- Fryns JP, Kleczkowska A, Timmermans J, van den Berghe H (October 1996). "The Floating-Harbor syndrome: two affected siblings in a family". Clinical Genetics. 50 (4): 217–9. doi:10.1111/j.1399-0004.1996.tb02629.x. PMID 9001802.
- Rosen AC, Newby RF, Sauer CM, Lacey T, Hammeke TA, Lubinsky MS (August 1998). "A further report on a case of Floating-Harbor Syndrome in a mother and daughter". Journal of Clinical and Experimental Neuropsychology. 20 (4): 483–95. doi:10.1076/jcen.20.4.483.1472. PMID 9892052.
- Ioan DM, Fryns JP (2003). "Floating-Harbor syndrome in two sisters: autosomal recessive inheritance or germinal mosaicism?". Genetic Counseling. 14 (4): 431–3. PMID 14738118.
- Hood RL, Lines MA, Nikkel SM, Schwartzentruber J, Beaulieu C, Nowaczyk MJ, et al. (February 2012). "Mutations in SRCAP, encoding SNF2-related CREBBP activator protein, cause Floating-Harbor syndrome". American Journal of Human Genetics. 90 (2): 308–13. doi:10.1016/j.ajhg.2011.12.001. PMC 3276662. PMID 22265015.
- Le Goff C, Mahaut C, Bottani A, Doray B, Goldenberg A, Moncla A, et al. (January 2013). "Not all floating-harbor syndrome cases are due to mutations in exon 34 of SRCAP". Human Mutation. 34 (1): 88–92. doi:10.1002/humu.22216. PMID 22965468.
- Robinson PL, Shohat M, Winter RM, Conte WJ, Gordon-Nesbitt D, Feingold M, et al. (October 1988). "A unique association of short stature, dysmorphic features, and speech impairment (Floating-Harbor syndrome)". The Journal of Pediatrics. 113 (4): 703–6. doi:10.1016/s0022-3476(88)80384-6. PMID 3171794.
- Hersh JH, Groom KR, Yen FF, Verdi GD (February 1998). "Changing phenotype in Floating-Harbor syndrome". American Journal of Medical Genetics. 76 (1): 58–61. doi:10.1002/(SICI)1096-8628(19980226)76:1<58::AID-AJMG10>3.0.CO;2-O. PMID 9508066.
- http://floatingharborsyndrome.co.uk/associated-difficultiesdisorders/microcephaly/
- Pelletier G, Feingold M (1973). "Case report 1. Syndrome identification". 1 (1): 8. Cite journal requires
|journal=(help) - Leisti J, Hollister DW, Rimoin DL (1975). "The Floating-Harbor syndrome". Birth Defects Original Article Series. 11 (5): 305. PMID 1218224.
- Feingold M (April 2006). "Thirty-two year follow-up of the first patient reported with the Floating-Harbor syndrome". American Journal of Medical Genetics. Part A. 140 (7): 782–4. doi:10.1002/ajmg.a.31159. PMID 16523514.
- García RJ, Kant SG, Wit JM, Mericq V (2012). "Clinical and genetic characteristics and effects of long-term growth hormone therapy in a girl with Floating-Harbor syndrome". Journal of Pediatric Endocrinology & Metabolism. 25 (1–2): 207–12. doi:10.1515/jpem.2011.406. PMID 22570979.
- Lopez E, Callier P, Cormier-Daire V, Lacombe D, Moncla A, Bottani A, et al. (February 2012). "Search for a gene responsible for Floating-Harbor syndrome on chromosome 12q15q21.1". American Journal of Medical Genetics. Part A. 158A (2): 333–9. doi:10.1002/ajmg.a.34401. PMID 22247066.
External links
| Classification | |
|---|---|
| External resources |
|