 |
|
|
|
|
|
|
| ||||||||||
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Creutzfeldt-Jakob Disease Not Related to a Common Venue --- New Jersey, 1995--2004Beginning in June 2003, the New Jersey Department of Health and Senior Services (NJDHSS) and CDC were notified of a suspected cluster of deaths caused by Creutzfeldt-Jakob disease (CJD) in persons reportedly linked to Garden State Racetrack in Cherry Hill, New Jersey. Concerns were raised that these deaths might have resulted from consumption of meat contaminated with the agent causing bovine spongiform encephalopathy (BSE, commonly called "mad cow disease") served at racetrack restaurants during 1988--1992. Consumption of BSE-contaminated cattle products has been linked to a new variant form of CJD (vCJD) in humans. This report summarizes the results of an investigation that determined the deaths were not linked causally to a common source of infection. The findings underscore the need for physicians to arrange for brain autopsies of all patients with clinically suspected or diagnosed CJD. Available clinical and neuropathologic findings were reviewed for 17 suspected CJD deaths referred to NJDHSS and CDC. To investigate the deaths of these 17 persons, all of whom were reportedly linked to Garden State Racetrack, health-care providers were contacted and relevant medical records obtained by NJDHSS, other state health departments, and CDC. Providers were asked to submit available brain autopsy tissue to the National Prion Disease Pathology Surveillance Center (NPDPSC), a national prion disease diagnostic referral laboratory established by CDC and the American Association of Neuropathologists. Sufficient demographic and clinical information was available to classify 11 of the 17 deaths as resulting from a definite or probable case of a classic form of CJD*, on the basis of World Health Organization criteria (1). Of the remaining six decedents, neuropathologic analyses documented that three deaths resulted from causes unrelated to either vCJD or classic CJD (Table 1). Three deaths reported as resulting from CJD remain under investigation. Excluding the three deaths for which CJD was ruled out, the 14 remaining deaths occurred over a period of approximately 9.25 years (1995--2004); the average number of cases per complete year (i.e., excluding 2004) was 1.44 (range: zero to three cases). Eleven of the 14 decedents were male; median age was 69.5 years (range: 50--83 years). Six of the decedents resided in New Jersey, four in Pennsylvania, and one each in Connecticut, Delaware, Maryland, and Virginia. Neuropathologic analysis in the five definite cases with available brain tissue specimens was diagnostic of classic CJD; none had the characteristic pathologic findings of vCJD. A genotype at codon 129 of the prion protein gene (a genetic marker associated with specific subtypes of CJD) was determined for three of the five CJD deaths confirmed pathologically (Table 1). None of the decedents had the methionine homozyogosity or the characteristic Western blot pattern present for persons with vCJD. In addition, the reported CJD subtypes differed from one another. For the six deaths without tissue diagnosis, available clinical and diagnostic evidence, including illness duration, electroencephalographic patterns, and presence of protein 14-3-3 (a marker for classic CJD) in cerebrospinal fluid was consistent with a probable diagnosis of classic CJD (Tables 1 and 2). None of the decedents had a diagnosis of vCJD. For 1995--2002, using CDC's national multiple cause-of-death file (2002 data are preliminary) compiled annually by the National Center for Health Statistics, the annual death rate from CJD in the United States has been stable at approximately one case per 1 million persons per year (Figure 1). The CJD death rate for New Jersey during the same period was similar. In 2001, Garden State Racetrack was closed permanently. The number and ages of all persons visiting or dining at the racetrack is unknown. However, according to New Jersey Racing Commission records, attendance at the racetrack during 1988--1992 was approximately 4.1 million. Based on an annual CJD rate of 3.4 cases per 1 million persons (CDC, unpublished data, 2004) and an overall death rate from all causes of 2.9% for persons aged >50 years, the occurrence over approximately 9.25 years (1995--2004) of at least 14 CJD-related deaths among as few as 300,000 persons aged >50 years would not be unusual. This number is within the estimated range of the number of persons attending and dining at the racetrack, given the known attendance. Reported by: P Gambetti, MD, National Prion Disease Pathology Surveillance Center, Case Western Reserve Univ, Cleveland, Ohio. J Hadler, MD, Connecticut Dept of Public Health. A Hathcock, PhD, M Drees, MD, Delaware Health and Social Svcs. D Blythe, MD, Maryland Dept of Health and Mental Hygiene. E Bresnitz, MD, M Gerwel, MD, New Jersey Dept of Health and Senior Svcs. M Hawkins, MD, Philadelphia Dept of Public Health; A Weltman, MD, Pennsylvania Dept of Health. J Marr, MD, A Buckler, MD, C Novak, MD, Virginia Health Dept. C Rothwell, MS, K Kochanek, MA, R Anderson, PhD, Div of Vital Statistics, National Center for Health Statistics; J Sejvar, MD, E Belay, MD, R Maddox, MPH, A Curns, MPH, R Holman, MS, L Schonberger, MD, Div of Viral and Rickettsial Diseases, National Center for Infectious Diseases, CDC. Editorial Note:CJD is a neurodegenerative disease characterized by rapidly progressive dementia associated with brain pathology marked by diffuse spongiform degeneration; the disease is invariably fatal (2). According to the leading hypothesis, CJD is caused by an unconventional transmissible agent, an abnormal protein (i.e., prion) that is able to induce abnormal folding of normal cellular proteins, leading to neuronal death. Prions are believed to cause transmissible spongiform encephalopathies (TSEs) that include scrapie in sheep, BSE in cattle, chronic wasting disease (CWD) in deer and elk, and CJD in humans. Two major forms of CJD have been recognized, classic and variant (3). Classic CJD has been recognized since the early 1920s and is characterized by certain distinct clinical and diagnostic features (Table 2). The most common form of classic CJD is believed to occur sporadically, caused by the spontaneous transformation of normal prion proteins into abnormal prions. This sporadic disease occurs worldwide at a rate of approximately one case per 1 million population per year, although rates of up to two cases per million are not unusual (4). Risk increases with age, and in persons aged >50 years, the annual rate is approximately 3.4 cases per million. Variant CJD was first described in 1996 in the United Kingdom and has different clinical characteristics than classic CJD (Table 2) (2,3). The median age at death for vCJD patients is 28 years, compared with 68 years for patients with classic CJD (Figure 2). In addition, all vCJD cases have neuropathologic findings distinctly different from those of classic CJD (5), and all have had a particular genetic profile (i.e., homozygosity for methionine) at codon 129 of the prion protein gene (4). Thus, cases of vCJD can be distinguished from classic CJD on the basis of clinical and pathologic data. Epidemiologic and laboratory evidence indicate that the agent causing BSE in cattle can be transmitted to humans via consumption of BSE-contaminated cattle products, causing vCJD (2,3). However, this evidence also suggests that the risk is low for having vCJD, even after consumption of contaminated product. In 1996, because of the emergence of vCJD in the United Kingdom, CDC enhanced its surveillance for CJD in the United States (6). No evidence has indicated that any of the 17 reported deaths resulted from vCJD. The CJD subtypes were determined in four decedents, and the subtype in each differed from the others; this heterogeneity provides scientific evidence against a common etiology for these cases. Although one study reported that BSE-infected mice expressing methionine homozygosity at codon 129 produced prions with a molecular phenotype consistent with a subtype of classic CJD (7), these animal data cannot be reliably extrapolated to humans in the absence of other supporting evidence. In 2003, the Spongiform Encephalopathy Advisory Committee of the United Kingdom concluded that these data did "not provide strong evidence to support" the hypothesis that exposure to BSE can produce a sporadic CJD-like phenotype in humans (8). In the United Kingdom, where the largest epidemic of BSE has occurred and an unusually large proportion of the population has been exposed to the BSE agent, the absence of an unusually high incidence of classic CJD patients or an elevated proportion of CJD patients with methionine homozygosity at codon 129 (9) supports the lack of association between BSE and sporadic CJD. In the United Kingdom, prion disease experts have looked specifically for evidence of BSE-related disease other than vCJD among classic CJD cases. No evidence of a new phenotype has been uncovered (R.G. Will, M.D., National CJD Surveillance Unit, Western General Hospital, Edinburgh, Scotland, personal communication, 2004). Neuropathologic evaluation, particularly by immunohistochemistry or Western blot, is the most definitive method to 1) diagnose human prion diseases, 2) monitor for vCJD and various subtypes of CJD, and 3) detect the possible emergence of new prion diseases in the United States. Although not all decedents in this investigation had pathologic specimens available for review, demonstration of the absence of a classic CJD or vCJD diagnosis in certain patients and diagnosis of classic CJD in others indicated these patients did not die from BSE-related disease. This investigation underscores the need for physicians to pursue autopsies of all decedents with clinically suspected and diagnosed CJD and to use the TSE diagnostic services provided free of charge by NPDPSC. Information regarding this surveillance center is available at http://www.cjdsurveillance.com or by telephone, 404-639-3091. CDC will continue to work with and support state health officials in New Jersey and nationally to conduct surveillance for CJD. Better defining the normal occurrence of subtypes of sporadic CJD and other TSEs will facilitate earlier recognition of vCJD or any other human prion disease that might emerge in the United States. * Those types of CJD that differ from vCJD and usually indicate sporadic CJD. References
Table 1 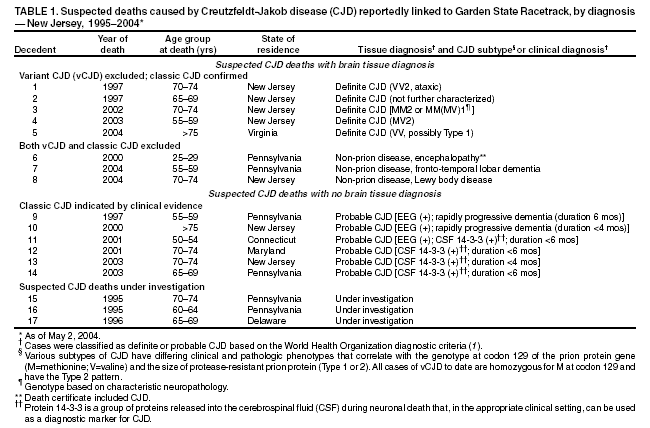 Return to top. Figure 1 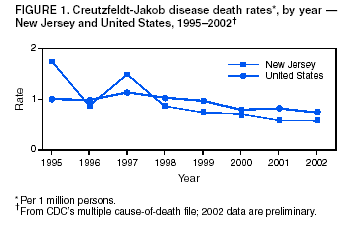 Return to top. Table 2  Return to top. Figure 2 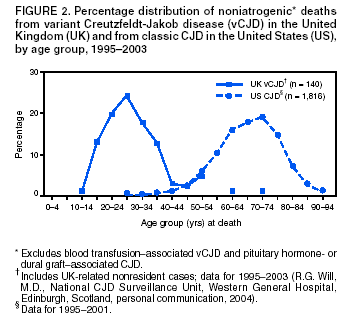 Return to top.
Disclaimer All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Page converted: 5/7/2004 |
|||||||||
This page last reviewed 5/7/2004
|