

Part IV: DEVELOPING, IMPLEMENTING, AND EVALUATING POPULATION INTERVENTIONS
 ShareCompartir
ShareCompartir
This website is archived for historical purposes and is no longer being maintained or updated.
necessarily represent the views of the funding agency.”
Genetics and Public Health in the 21st Century
-
Chapter 18
Genetics and Prevention Effectiveness -
Chapter 22
Newborn Screening for Sickle Cell Disease: Public Health Impact and Evaluation - Chapter 23
Public Health Strategies to Prevent the Complications of Hemochromatosis
Public Health Strategies to Prevent the Complications of Hemochromatosis
Wylie Burke1 , Mary E. Cogswell 2, Sharon M. McDonnell2, and Adele Franks3
1Department of Medicine, University of Washington, Seattle, WA98105
2Division of Nutrition and Physical Activity, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention, 3005 Chamblee-Tucker Road, Atlanta, GA30341
3Prudential Center for Healthcare Research, 2859 Paces Ferry Road, N.E., Suite 820, Atlanta, GA30339
INTRODUCTION
Hemochromatosis is a treatable adult-onset genetic disorder for which screening tests are available. These characteristics make hemochromatosis an important model for public health to explore as it begins to address genetic contributors to adult disease. Whereas newborn screening programs are motivated by the need to provide early treatment of genetic conditions like phenylketonuria (PKU), this urgency does not apply to adult-onset disorders like hemochromatosis. Conversely, other adult onset disorders for which genetic testing is available, such as hereditary breast/ovarian cancer and Alzheimer disease, are not readily treatable.As our knowledge of genetic susceptibility to chronic diseases grows, the issues raised in the public health approach tohemochromatosis are likely to be relevant to many future efforts in genetics and public health.
Hemochromatosis results in the accumulation of excess iron stores over time. Complications occur when iron overload is sufficient to cause organ damage; these complications include cirrhosis, primary liver cancer, cardiomyopathy, arthritis and diabetes.[1-3] Current therapy for hemochromatosis starts with a “de-ironing” procedure at diagnosis, also called quantitative phlebotomy. This procedure consists of the removal of multiple units of blood, often over a period of several months.After the initial therapy, a maintenance
program of periodic blood drawing is instituted, to prevent re-accumulation of iron stores. [1-3] This therapy prolongs survival in symptomatic persons and appears to normalize life expectancy if begun early in the course of the disease. [4]
Accordingly, early diagnosis of hemochromatosis can be expected to reduce the burden of the disease. A decrease in the proportion of affected persons with late-stage disease has already been seen in recently reported clinical cohorts, compared with those reported two or three decades ago. [3-5] This shift reflects changing criteria for diagnosis, and in particular, increased use of abnormal serum iron measures such as elevated transferrin saturation or serum ferritin to detect persons with iron overload. [3,5] Even in these recent reports, however, a significant number of affected persons had diabetes, heart disease or cirrhosis at the time of diagnosis. [3,5] Increasing physician awareness of the need for early diagnosis and treatment of hemochromatosis could result in further reductions in late-stage complications. We call this approach to the prevention of iron overload disease enhanced case finding.
Another strategy for reducing disease burden is universal screening. Serum iron measures – in particular transferrin saturation (TS = serum iron/total iron binding capacity X 100) – can be used to identify asymptomatic persons with iron overload.[6] DNA-based tests for mutations in the HFE gene also provide a means to detect asymptomatic persons. [7] Cost analyses suggest that the cost of TS screening is offset by savings from reduced end-stage complications of hemochromatosis.[8-10] DNA-based screening might also be cost-effective, if the sensitivity of the test were high and the cost of testing could be reduced to a level similar to TS testing. [11]
An elevated TS value or a positive result on a genetic test, however, does not predict certain progression to symptoms or serious complications of hemochromatosis. It is not known what proportion of persons with a positive screening test will remain healthy without treatment. Nor is it known whether the full benefits of treatment require detection at an asymptomatic stage, as opposed to detection at the time of early signs or symptoms. Thus, calculations of the cost-effectiveness of screening for hemochromatosis are to some degree speculative. In addition, little attention has been given to the resources required to implement and maintain hemochromatosis screening programs, or to the social or economic effects of screening. Whether these uncertainties provide a substantive rationale against screening has been the subject of debate. [12]
Enhanced case finding can be seen as the first stage in a public health response, when evidence for an effective early treatment of a disorder has emerged.In contrast, universal screening generally calls for more stringent evidence of benefit than case finding, because screening involves the testing and treatment of healthy people who are without medical complaints. In this chapter we review current knowledge about the natural history and genetics of hemochromatosis, and consider the implications for public health policy of a transition from enhanced case finding to universal screening.
Uncertainties about the Natural History and Disease Burden of Hemochromatosis
For both enhanced case finding and universal screening, a reliable means of diagnosing hemochromatosis at an early stage is needed. Persistently elevated TS – defined as an elevated TS value on both a random blood draw and a follow-up fasting sample – is an indicator of possible hemochromatosis. The serum ferritin level can also be used to detect affected persons in an asymptomatic stage. However, the serum ferritin level is less sensitive than TS, because it usually rises later in the disease process, and is also less specific because it is elevated in other common conditions, such as infection and inflammation. [6] Some experts recommend using elevated serum ferritin as an indicator for liver biopsy, to determine whether cirrhosis is present or not, after a persistently elevated TS has been documented. [3,6]
TS screening studies have used thresholds ranging from 45% to 70% to define an elevated TS value. [12-15] The percentage of the US white population with an elevated random TS ranges from 1% to 6%, depending on both the threshold used and the population tested [10, 13-16]; of these 10-35% will have a persistently elevated TS value on a follow-up fasting test. [13-15] Among those with persistently elevated TS, 40% to 75% will have evidence of iron overload by liver biopsy or quantitative phlebotomy [13-15]; the remainder are likely to include some affected persons in the early stages of iron accumulation. Estimates of the prevalence of hemochromatosis derived from TS screening studies range from 2 to 8 per 1000. [8-11,12-15]
Yet rates of documented hospitalizations, outpatient visits, and mortality attributed to hemochromatosis are much lower than predicted by TS screening studies. [12,17] This discrepancy suggests that hemochromatosis is underdiagnosed, or that the risk of morbidity and mortality among persons identified through TS screening is low, or both.[12] Several factors may contribute to underdiagnosis of hemochromatosis.Methods of documentation may contribute; for example,some data sources (such as, Medicaid data) are limited to one diagnosis. If a complication of hemochromatosis is the primary reason for a visit (e.g., diabetes mellitus, chronic liver disease, or arthritis), hemochromatosis is unlikely to be documented. Failure to recognize hemochromatosis may also contribute to underdiagnosis. Until recently, the diagnosis of hemochromatosis relied on late-stage symptoms. One patient survey indicated that many affected persons experience a long delay between the onset of symptoms and the diagnosis of hemochromatosis, even when medical consultation is sought repeatedly. [18] Screening studies among persons with chronic disease such as arthritis and diabetes also suggest that when hemochromatosis is the underlying cause of the disease it is often unrecognized [12,19-21].
Although failure to diagnose hemochromatosis is a likely contributor to the low rate of documented cases, lack of disease progression among persons who would be identified in screening programs also seems likely. In large population screening studies, only 45% of men and 43% of women aged >40 years with iron overload exhibited one or more clinical manifestations of hemochromatosis. [22] These percentages are high enough to predict many more cases of iron overload than are seen in clinical data sources. The clinical manifestations assessed in these large screening studies, however, included both the severe complications of hemochromatosis (such as liver disease, diabetes, and cardiomyopathy) and the more nonspecific symptoms seen early in the course of the disease (arthropathy,fatigue, weight loss, abdominal pain, and impotence). [22] Because the nonspecific symptoms are relatively common among persons without hemochromatosis, some of the morbidity attributed to hemochromatosis in screening studies could be due to other causes. It is thus possible that only a subset of persons detected in screening programs would develop disorders attributable to iron overload.
No screening studies have reported longitudinal follow-up. The current standard of care for persons with persistently elevated TS (in the absence of other causes) is to proceed to quantitative phlebotomy, both for confirmation of the diagnosis and to initiate therapy. In addition, serum ferritin and liver function tests are usually done, and a liver biopsy recommended if the results of these tests are significantly elevated. Ongoing blood removal is recommended if quantitative phlebotomy or liver biopsy indicates iron overload. [1,3] Because TS screening leads to treatment, prospective follow-up in itself cannot be used to determine the proportion of persons testing positive who would remain healthy over time if untreated.
Contribution of Genetic Testing to the Diagnosis of Hemochromatosis
The discovery of the gene for hemochromatosis, HFE, [23,24] was expected to provide a clearer approach to diagnosis. Genetic testing, however, has turned out to involve uncertainties similar to those posed by TS testing. Two HFEmutations have been defined, C282Y and H63D. [23] In control populations, carriers of the H63D mutation are more common thanC282Y carriers. [23,25-33] In genotype studies of persons with hemochromatosis, however, most affected persons are homozygous for the C282Y mutation (60%-100%).[23, 25-33] A small proportion are compound heterozygotes (C282Y/H63D) (0%-7%) or H63D homozygotes (0%-4%). [23, 25-33] Some persons with iron overload are heterozygous for one of the mutations (0%-15%) or have no identifiable HFEmutation (0%-21%), suggesting that as yet unidentified mutations may contribute to the etiology of hemochromatosis.[23,25-33]
These data indicate that the C282Y mutation is associated with a higher risk of iron overload than the H63D mutation, a conclusion supported by a pooled analysis of case-control studies reporting HFEgenotype data [34] and by functional studies of the HFEprotein. [35-37] The pooled analysis indicated a gradient of risk for iron overload according to HFEgenotype: risk was much higher in C282Y homozygotes than in persons with other HFEgenotypes, and was progressively lower for C282Y/H63D, H63D/H63D and C282Y heterozygosity.[34,38] A model for this gradient of risk is illustrated in Figure 1. Taken together, the genetic data indicate that DNA-based testing cannot provide a simple positive test result for hemochromatosis, but instead identifies many persons with a low or intermediate risk of iron overload. Even among C282Y homozygotes, risk of future disease is uncertain. Case reports have documented elderly persons with this genotype who are without evidence of significant disease; some have no evidence of iron overload. [3,31,39]
Figure 1
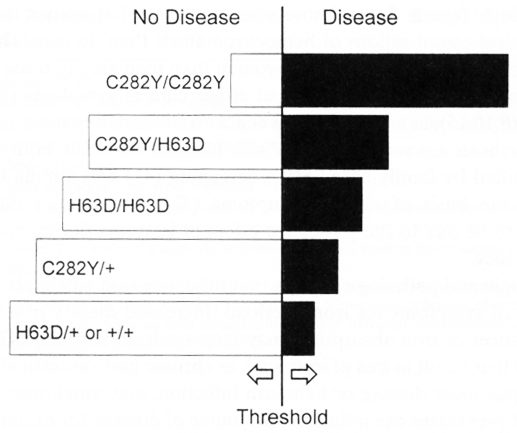
Figure 23.1 The likelihood of developing overt hemochromatosis is shown as a function of HFE genotype. C282Y homozygotes have the highest risk, with risk declining progressively for compound heterozygotes, H63D homozygotes, and C282Y heterozgotes.H63D heterozygotes are assumed to have the same minimal degree of risk as persons without identifiable mutations; in this category, the risk reflects the small possibility of carrying mutations not currently identifiable. For all genotypes, the risk of disease increases or decreases according to the presence of modifying factors such as gender, alcohol use and iron intake.
Risk Modifiers
Interacting with the susceptibility conferred by HFE mutations are modifiers that contribute to the risk of iron overload, such as gender and physiologic and pathologic factors. Several observations suggest that women have a lower risk of clinical complications of hemochromatosis than men. These observations include the finding of fewer women than men among persons diagnosed with hemochromatosis on the basis of either clinical symptoms [4,40] or TS screening. [8,10,15] In addition, lower concentrations of hepatic iron and fewer cases of cirrhosis are seen in women with hemochromatosis, both among siblings identified by family-based HLA screening [41] and among persons diagnosed on the basis of clinical symptoms. [42] This gender difference is presumed to be due to the protective effect of iron loss during menstruation and pregnancy.
Physiologic and pathologic factors that influence iron stores also affect the likelihood of symptoms for iron overload. Increased dietary iron or vitamin C, an enhancer of iron absorption, may increase iron overload [43], whereas conditions that result in loss of iron, such as chronic gastrointestinal blood loss due to peptic ulcer disease or helminth infection, may ameliorate iron overload.[43] Liver toxins can influence the course of disease; for example,alcohol abuse increases the likelihood of liver disease and is associated with decreased survival among persons with hemochromatosis. [33,44,45] Chronic hepatitis may be also a contributor to liver disease in hemochromatosis. [33] In addition to increased absorption of iron, increased absorption of zinc and lead has been observed in patients with hemochromatosis [46,47] and may contribute to end-stage organ damage.
What meaning should be applied to a diagnosis of hemochromatosis?
Several methods are available for identifying persons in the early stages of hemochromatosis, as summarized in Table 1. For each method, choices must be made concerning the cutoffs used to define a positive test result. All methods potentially generate false-positive and false-negative results, but the sensitivity, specificity, and predictive value of any given method can be calculated only after there is agreement about the best case definition. The lack of a single accepted case definition for early-stage hemochromatosis represents the major difficulty in evaluating methods for early detection.
Table 1
Potential Case Definitions for the Early Diagnosis of Hemochromatosis |
|---|
| Persistently elevated TS1 |
| Persistently elevated TS1and iron overload2 |
| Persistently elevated TS1and iron overload2 and symptoms or signs of hemochormatosis |
| Persistently elevated TS1and hemochromatosis genotype3 |
| Hemochromatosis genotype3 |
| Hemochromatosis genotype3and iron overload2 |
| Hemochromatosis genotype3and iron overload2 and symptoms or signs of hemochormatosis |
1. Persistently elevated TS = Elevated TS level on a random blood sample and a follow-up fasting blood sample
2. Iron overload = Elevated hepatic iron index (>1.9) on tissue sample from liver biopsy,or > 4 g or iron removed by quantitative phlebotomy.The latter is a series of blood draws that allows a quantification of iron stores.1-2 units of blood are drawn each week until serum ferritin is below 100 ng/mL or hematocrit is below normal
3. Hemochromatosis genotype = Presence of two HFE mutations.
In considering what should constitute a diagnosis of hemochromatosis, the meaning applied to the diagnosis must be taken into account. Should hemochromatosis be considered a risk state, akin to hypertension or hypercholesterolemia? Or is it more accurately classified as a genetic disease with delayed but potentially severe complications, like PKU? In the usual sense of the term, a genetic disease carries with it the implication that all or most persons carrying the disease-associated genotype will be clinically affected. In this context, a genetic test is assumed to be highly predictive of future disease. The differential risk of disease seen with different HFEgenotypes, including evidence of incomplete penetrance for the genotype conferring the highest risk , [3,31,39] makes it difficult to define hemochromatosis as a genetic disease in this traditional sense.
Another approach is to consider hemochromatosis as a genetic susceptibility state rather than as a disease condition. This distinction is more than semantic because it has implications for case definition, for the other diagnoses with which hemochromatosis should be compared, and for patients’ and providers’ expectations after a diagnosis is made. When hemochromatosis is defined by a simple inclusive criterion – for example, persistently elevated TS, in the absence of other causes -a pool of people with increased risk can be readily identified. Such a definition is comparable to identifying hypertensive or hypercholesterolemic persons on the basis of blood pressure or serum cholesterol level. Treatment may benefit only a subset of persons with persistently elevated TS, but the same is true of mild to moderate hypertension and hypercholesterolemia: the absolute reduction in cardiovascular risk produced by treatment of elevated blood pressure or cholesterol is small. [48] Many persons are labeled as hypertensive or hypercholesterolemic so that a minority can avoid premature cardiovascular events [48]; this is done because there is no method for determining which persons with elevated blood pressure or hypercholesterolemia will suffer adverse events if untreated.Identification of persons with persistently elevated TS may provide a similar benefit.
Enhanced Case Finding
The purpose of enhanced case finding is to reduce the morbidity and mortality of hemochromatosis through early detection and treatment of affected persons in clinical settings. This approach assumes that detecting hemochromatosis at the time symptoms first appear is sufficient to gain the benefits of phlebotomy treatment. Although controlled trials of phlebotomy treatment have not been conducted, observational data provide strong support for its efficacy in reducing mortality. These data include the documentation of an apparently normal life expectancy in persons treated before the onset of cirrhosis or diabetes [4,49] and a prolongation of survival in patients at all stages of the disease compared with historic controls.[4,50] The efficacy of phlebotomy treatment in reducing early symptoms such as fatigue or joint pain is unknown.
The initiation of enhanced case finding would require a concerted educational campaign to increase awareness of hemochromatosis among physicians and health care systems. [51] Key educational points to be included in he educational campaign are: the estimated prevalence of hemochromatosis, the severity of end-stage organ complications, the efficacy of treatment, and the nature of the common nonspecific symptoms seen early in the course of disease. In addition, physicians are likely to benefit from guidance on the use and interpretation of serum iron measures and other diagnostic strategies for hemochromatosis.
Educational efforts are likely to succeed only if they move beyond conventional conference-based continuing medical education to incorporate practice-based interventions. [52] Approaches such as adding TS testing to the usual work-up of patients with newly diagnosed diabetes, arthritis and impotence, or of patients with ill-defined abdominal pain or fatigue, may be helpful. In addition to an educational effort for health care providers, this approach may require public education, to increase the likelihood that complaints consistent with early hemochromatosis are evaluated.[51]
A program to increase the use of TS testing would also require efforts to ensure laboratory standardization and quality assurance. Multiple laboratory methods are available for determining serum iron and total iron-binding capacity (the components of a TS measurement); a pilot study of laboratory proficiency found significant variability in these measurements. [53] Standardization procedures, including the development of an analytic reference system similar to that implemented for serum cholesterol, may be required to ensure an acceptable level of accuracy for TS measurements. [13,53] The same issues of standardization apply to genetic testing as well.
The enhanced case finding approach has several advantages.Because testing occurs within the usual health care setting, no additional screening infrastructure is required, and follow-up is accessible to those with positive test results. Although a substantial proportion of patients seen in primary care settings may have complaints that merit consideration of testing for hemochromatosis, the number tested will still be considerably lower than would be the case with universal screening. In addition, testing will be done in the context of an identified health problem. These factors would reduce the number of persons with a false-positive test result.Further, when a diagnosis of hemochromatosis is made, it will occur in the context of a patient seeking care and thus will avoid diagnostic labeling of persons who consider themselves to be healthy.
In contrast, several factors may limit the benefits of enhanced case finding. Persons with early symptoms may not be evaluated or treated, and appropriate treatment may not occur after the diagnosis is made. Lack of adequate follow-up has been documented in other early detection programs[54,55]; any educational efforts or quality assurance programs developed to increase detection of hemochromatosis should include efforts to monitor the effectiveness of medical follow-up.
The biggest potential drawback to this approach, however, is the possibility that treatment initiated after symptoms occur may be too late to achieve the full benefits of prevention. Even if treatment on the basis of early symptoms is sufficient to prevent premature mortality, patients’ quality of life could be diminished if early symptoms are not reversible. These considerations underscore the need for a systematic study of the natural history of hemochromatosis. In addition, data are needed on the efficacy of phlebotomy treatment in reversing early symptoms of hemochromatosis.
Universal Screening
Compared with enhanced case finding, universal screening for hemochromatosis would lead to more testing and more false-positive results, but would also offer the possibility of a greater reduction in the avoidable medical complications of hemochromatosis. The primary assumption underlying universal screening is that hemochromatosis must be detected before symptoms occur to fully prevent the complications of the iron overload. Proponents of universal screening also assume that the risks and harms of screening asymptomatic persons are outweighed by the benefits of early detection and treatment.
Implementation of universal screening would require additional resources within the health care system, the nature of which would depend on the strategy chosen to accomplish screening. If screening is proposed as an adjunct to other routine health care, for example, offered at the same time as Pap testing or colorectal cancer screening, the resources required would be related primarily to health provider education, similar to what would be required for enhanced case finding. For men, however, screening in early adulthood might be difficult to accomplish in this way, because fewer men than women are likely to be regular users of preventive health care services between 20 and 50. If separate screening programs were instituted, resources for a screening infrastructure would be needed, including provisions to ensure follow-up care of those who screen positive. Laboratory standardization issues would be of the same nature as for case finding, but on a larger scale.If screening included genetic testing, current practice standards would require pre- and posttest genetic counseling as well.
With the larger number of persons screened and treated, universal screening increases the number of persons exposed to the possibility of adverse psychologic, social or economic consequences of a diagnosis of hemochromatosis. The potential for loss of health insurance and employment after a genetic diagnosis is a concern for both consumers and policymakers [56-59]. Legislative efforts to minimize these risks are being implemented [60], but the degree of protection they will provide is unknown. Although adverse outcomes after a diagnosis of hemochromatosis have been reported [18,61], no systematic study has been undertaken to assess them.
Adverse psychologic effects of screening may occur, but the few data available for other conditions are conflicting. Increased absenteeism from work has been seen after a diagnosis of hypertension, [62] and perceptions of ill health have been described after a diagnosis of hypercholesterolemia [63]. Conversely, a workplace cholesterol screening trial and a primary care-based assessment of cardiovascular risk factors failed to show evidence of adverse psychologic effects by standard measures. [64,65] The wide acceptance of these screening strategies implies that many view the benefits of cardiovascular screening as outweighing any psychologic burdens imposed. There may be greater reluctance to proceed with screening for a genetic condition than with screening for non-genetic risk factors, however, because the personal burdens are perceived as being heavier. [66] These include the ramifications of identifying a risk that may affect family members and, perhaps more important, the possibility that a genetic diagnosis may be stigmatizing. [66] The way in which a disorder is understood may influence the psychological response to a positive screening test. [64,67].With hemochromatosis, the psychologic burdens of the diagnosis may be reduced by clear communication that it is an indicator of increased susceptibility rather than a prediction of certain future disease, and that treatment can be offered to substantially reduce the risk of developing disease. Whether communication of this kind can change the perception of hemochromatosis from a genetic disease to a risk state, or reduce the likelihood of discrimination, remains to be determined. More research is needed to address these questions.
Conclusions
Enhanced case finding can be justified on the basis of current evidence, because a reasonable likelihood of benefit can be inferred when symptomatic persons are treated. Thus, efforts to increase public and health care provider awareness of hemochromatosis are merited. The educational programs required to increase the early detection of hemochromatosis could also serve as a preparatory step in the development of universal screening programs.
The strongest argument in favor of universal screening is the possibility that enhanced case finding will fail to detect affected persons before irreversible complications of hemochromatosis occur. However, reductions in the frequency of late-stage complications of hemochromatosis appear to be occurring already, in the absence of large-scale programs to improve case finding. Thus, more direct evidence for outcome benefit is needed to justify universal screening of healthy persons. This is particularly true given the uncertainty about the number of persons who might be exposed to unnecessary treatment and the unknown potential for social, psychologic and economic harms, if universal screening were implemented.
The burden of disease associated with hemochromatosis also needs to be considered when addressing the question of universal screening. Although hemochromatosis can be construed as a risk state similar to hypertension and hypercholesterolemia, it is much less common than these diagnoses (approximately 15% of adults are hypertensive [48] and 20% are hypercholesterolemic, [68] with the proportion rising with age for both). Hemochromatosis is thus a less common contributor to the disease burden of the population than either hypertension or hypercholesterolemia, and the resources required for universal screening may not be merited on this basis.
In determining the value of universal screening, however, the prevalence of the disorder may be less important than the effectiveness of screening and treatment. The two conditions for which newborn screening is mandated throughout the United States, PKU and congenital hypothyroidism, occur in about 1 in 12,000 and 1 in 3600 newborns, respectively.[48] Universal newborn screening is recommended, despite the low prevalence of these conditions, because of the effectiveness of screening and subsequent treatment in preventing mental retardation and other neurological complications inaffected infants. [48] These examples highlight timing of treatment as an important factor in determining the need for screening, that is, whether therapy must be initiated before symptoms occur to provide benefit. For PKU or neonatal hypothyroidism, treatment of asymptomatic infants is essential if complications are to be avoided. In contrast, when a mildly symptomatic latency period occurs – as in adult onset hypothyroidism, for example – efforts to increase case finding are likely to be more efficient for improving health outcome than a policy of universal screening.
The data critical to a consideration of universal screening for hemochromatosis thus include a full description of the symptoms associated with the early stages of the disease, their reversibility, and the degree to which other complications are prevented when treatment is initiated early.In addition, more information is needed on the potential for personal and economic harms resulting from a diagnosis of hemochromatosis and the measures available to reduce or prevent such risks. One important way to minimize the risk of adverse labeling is to limit the diagnosis of hemochromatosis to those most likely to benefit from treatment.Better information about the natural history of hemochromatosis is needed to accomplish this goal.
If hemochromatosis is underdiagnosed, as current estimates of prevalence suggest, much of the data needed on the natural history of the disease may be obtainable with well-designed case-control and cross-sectional studies that include an adequate sample of older persons. Such research should be able to define the disease status by age of persons who carry HFEmutations or have biochemical evidence of iron overload, and should allow a better estimation of the proportion of young asymptomatic persons likely to benefit from therapy. If these data supported screening, pilot studies that include assessment of the psychosocial outcomes of screening would be merited. As a matter of policy, it is important to ensure that evidence of this kind is generated before screening options are evaluated further. Without such data, the added benefit provided by universal screening cannot be determined.
The issues raised in the consideration of universal screening for hemochromatosis are likely to be relevant to future discussions of genetics and public health. Many new genetic tests emerging from current research are related to diseases of adult onset, and most identify an increased probability of disease, rather than a certainty. Because future disease is not certain, the effect of interventions designed to reduce risk may be difficult to measure. Yet the value of testing can be determined only by weighing the effectiveness of screening and treatment against potential psychological, social and economic risks.As the hemochromatosis example illustrates, these questions constitute a research agenda.
In the translation of genetic advances into effective public health action, research strategies that address the multiple aspects of genetic susceptibility are needed.As other tests to identify genetically susceptible persons are developed, targeted interventions to reduce risk must be developed and assessed. This effort will require knowledge about the natural history of the genetic condition, including the gene-environment interactions that influence disease risk and response to treatment. Evaluation of the outcomes of genetic testing will also require an assessment of the psychosocial consequences of testing and treatment. Hemochromatosis provides an early and instructive example of the interdependence of these important questions.
REFERENCES:
- Bothwell TH, Charlton RW, Motulsky AG. Hemochromatosis. In eds. Scriver CR,Beaudet AL, Sly WS, Valle D; The Metabolic and Molecular Bases of Inherited Disease, 7th ed.New York, NY: McGraw Hill; 1995.
- Motulsky AG, Wolff RK. Update on hemochromatosis, addendum to chapter 69 In eds. Scriver CR,Beaudet AL, Sly WS, Valle D; The Metabolic and Molecular Bases of Inherited Disease[book in CD-ROM]. New York, NY: McGraw Hill; 1997.
- Bacon BR. Diagnosis and management of hemochromatosis. Gastroenterology 1997; 113:995-999.
- Niederau C, Fischer R, Purschel A, et al. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 1996;110: 1107-19.
- Adams PC, Valberg LS. Evolving expression of hereditary hemochromatosis. Semin Liver Dis 1996; 16:47-54.
- Witte DL, Crosby WH, Edwards CQ, Fairbanks VF, Mitros FA. Practice guideline development task force of the College of American Pathologists. Hereditary hemochromatosis. Clin Chim Acta; 1996; 245:139-200.
- Burke W, Thomson E, Khoury MJ et al. Hereditary hemochromatosis: gene discovery and its implications for population-based screening. JAMA 1998; 280: 172-178.
- Balan V, Baldus W, Fairbanks V, Michels V, Burritt M, Klee G. Screening for hemochromatosis: cost-effectiveness study based on 12,258 patients. Gastroenterology 1994; 107:453-459.
- Phatak PD, Guzman G, Woll JE, et al. Cost-effectiveness of screening for hereditary hemochromatosis. Arch Intern Med 1994;154:769-776.
- Edwards CQ, Griffen LM, Goldgar D et al. Prevalence of hemochromatosis among 11,065 presumably healthy blood donors. N Engl J Med 1988; 318: 1355-1362.
- Adams PC, Valberg LS.Screening blood donors for hemochromatosis: decision analysis comparing genotypeing to phenotyping. Gastroenterology 1997; 112: 1207.
- Cogswell ME, McDonnell SM, Khoury MJ, Franks AL, Burke W, Brittenham G. Iron overload, public health and genetics: evaluating the evidence for hemochromatosis screening. Ann Intern Med 1998; 129: 971-979.
- McDonnell SM, Phatak PD, Felitti V, Hover A, McLaren GD. Screening for hemochromatosis in primary care. Ann Intern Med 1998; 129: 962-970.
- Adams PC, Gregor JC, Kertesz AE, Valberg LS. Screening blood donors for hereditary hemochromatosis: Decision analysis model based on a 30 year database. Gastroenterology 1995; 109:177-186.
- Phatak PD, Sham RL, Raubertas RF et al. Prevalence of hereditary hemochromatosis in a sample of 16,031 primary care patients. Ann Intern Med 1998; 129: 954-961.
- Looker AC, Johnson CL. Prevalence of elevated serum transferrin saturation in adults in the United States. Ann Intern Med 1998; 129:940-945.
- Yang Q, McDonnell SM, Khoury MJ, Cono J, Parrish RG. Hemochromatosis-associated mortality in the United States from 1979 to 1992: an analysis of multiple-cause mortality data. Ann Intern Med 1998; 129: 946-953.
- McDonnell SM, Preston BL, Jewell SA et al. A survey of patients with hemochromatosis: symptoms and response to treatment. Am J Med, in press.
- Conte D, Manachino D, Colli A et al. Prevalence of genetic hemochromatosis in a cohort of Italian patients with diabetes mellitus. Ann Intern Med 1998; 128: 370-373.
- Olynyk J, Hall P, Ahern M, Kwiatek R, Mackinnon M. Screening for genetic haemochromatosis in a rheumatology clinic. Aust N Z J Med. 1994; 24:588-589
- George DK, Evans RM, Crofton RW, Gunn IR. Testing for haemochromatosis in the diabetic clinic. Ann Clin Biochem. 1995;32:521-526.
- Bradley LA, Haddow JE, Palomaki GE. Population screening for haemochromatosis:a unifying analysis of published intervention trials. J Med Screen. 1996;3:178-184.
- Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996;13:399-408.
- Mercier B, Mura C, Ferec C et al., on behalf of the WHO Nomenclature Committee for Factors of the HLA System. Putting a hold on “HLA-H.” Nat Genet 1997;15(3):234..
- Beutler E, Gelbart T, West C et al. Mutation analysis in hereditary hemochromatosis. Blood Cells Mol Dis 1996;22:187-194.
- Barton JC, Shih WWH, Sawada-Hirai R et al. Genetic and clinical description of hemochromatosis probands and heterozygotes: evidence that multiple genes linked to the major histocompatibility complex are responsible for hemochromatosis. Blood Cells Mol Dis 1997; 23:135-145.
- Carella M, D’Ambrosio L, Totaro A et al. Mutation analysis of the HFE gene in Italian hemochromatosis patients. Am J Hum Genet 1997; 60:828-832.
- Jazwinska EC, Cullen LM, Busfield F et al. Hemochromatosis and HLA-H. Nat Genet 1996; 14:249-251.
- 29. Jouanolle AM, Gandon G, Jezequel P et al. Hemochromatosis and HLA-H. Nat Genet 1996; 14:251-252.
- Borot N, Roth M-P, Malfroy L et al. Mutations in the MHC class 1-like candidate gene for hemochromatosis in French patients. Immunogenetics1997;45:320-324.
- Adams PC, Chakrabarti S. Genotypic/phenotypic correlations in genetic hemochromatosis: evolution of diagnostic criteria. Gastroenterology 1998; 114: 319-323.
- UK Haemochromatosis Consortium. A simple genetic test identifies 90 percent of UK patients with
haemochromatosis. Gut 1997; 41: 841-844. - Piperno A, Sampietro M, Pietrangelo A et al. Heterogeneity of hemochromatosis in Italy. Gastroenterology 1998; 114: 996-1002.
- Burke W, McDonnell SM, Khoury MK.Contribution of different genotypes in the HFE gene to the etiology of hemochromatosis: a pooled analysis. Presented at the 13th National Conference on Chronic Disease Prevention and Control. Atlanta GA, Dec 8, 1998.
- Feder JN, Tsuchiihashi Z, Irrinki A et al. The hemochromatosis founder mutation in HLA-H disrupts b2-macroglobulin interaction and cell surface expression. J Biol Chem 1997; 272: 14025-14028.
- Waheed A, Parkkila S, Zhou XY et al. Hereditary hemochromatosis: effects of C282Y and H63D mutations on association with b2-macroglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells. Proc Natl Acad Sci USA 1997; 94: 12384-12389.
- Feder JN, Penny DM, Irrinki A et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA 1998; 95: 1472-1477.
- Burke W, Press N, McDonnell SM. Hemochromatosis: genetics helps to define a multifactorial disease. Clin Genet 1998; 54: 1-9.
- Adams PC, Campion ML, Gardon G et al. Clinical and family studies in genetics hemochromatosis: microsatellite and HFE studies in five atypical famlies. Hepatology 1997; 26:991-995.
- Adams PC, Deugnier Y, Moirand R, Brissot P. The relationship between iron overload, clinical symptoms and age in 410 hemochromatosis patients. Hepatology 1997; 25: 162-166.
- Edwards CQ, Griffen LM, Kushner JP. The morbidity of hemochromatosis among clinically unselected homozygotes: preliminary report. Adv Exp Med Biol 1994;356:303-308.
- Moirand R, Adams PC, Bicheler V, Brissot P, Deugnier Y. Clinical features of genetic hemochromatosis in womencompared to men. Ann Intern Med 1997;127:105-110.
- Powell LW, Burt MJ, Halliday JW, Jazwinska EC. Hemochromatosis: genetics and pathogenesis. Semin Liver Dis 1996; 16:55-63.
- Adams PC, Agnew S. Alcoholism in hereditary hemochromatosis revisited: prevalence and clinical consequences amongst homozygous siblings. Hepatology 1996; 23: 724-727.
- Loreal O, Duegnier Y, Morand R et al. Liver fibrosis in genetic hemochromatosis. Respective roles of iron and non-ironrelated factors in 127 homozygous patients. J Hepatol 1992; 16: 122-127.
- Adams PC, Bradley C, Frei JV. Hepatic zinc in hemochromatosis. Clin Invest Med 1991; 141:16-20.
- Barton JC, Patton MA, Edwards CQ et al. Blood lead concentrations in hereditary hemochromatosis. J Lab Clin Med 1994; 124: 193-198.
- US Preventive Service Task Force.Guide to Clinical Preventive Services, 2nd ediiton. Baltimore, MD; Williams and Wilkins; 1996.
- Adams PC, Speechley M, Kertesz AE. Long-term survival analysis in hereditaryhemochromatosis. Gastroenterology. 1991;101:368-372.
- Bomford A, Williams R. Long term results of venesection therapy in idiopathic hemochromatosis. Q J Med. 1976;45:611-623.
- NcDonnell SM, Witte DL, Cogswell ME, McIntyre R. Strategies to increase dtection of hemochromatosis. Ann Intern Med 1998; 129: 987-992.
- Davis DA, Thomson MA, Oxman AD, Haynes RB. Changing physician performance. JAMA 1995; 274: 700-705.
- Elaine Gunter, personal communication.
- Bonelli L, Branca M, Ferreri M, Barizzone D, Rossi E et al. Attitude of women toward early cancer detection and estimation of compliance to a screening program for cervix and breast cancer. Cancer Detect Prev 1996; 20: 342-352.
- Paskett ED, McMahon K, Tatum C, Velez C, Shelton B et al. Clinic-based interventions to promote breast and cervical cancer screening. Prev Med 1998; 27: 120-128.
- Hudson KL, Rothenberg KH, Andrews LB, Kahn MJE, Collins FS. Genetic discrimination and health insurance: an urgent need for reform. Science 1995; 270:391-393.
- Lapham EV, Kozma C, Weiss JO.Genetic discrimination: perspectives of consumers. Science 1996; 274: 621-624.
- Rothenberg K, Fuller B, Rothstien M et al.Genetic information and the workplace: legislative approaches and policy challenges. Science 1997: 275: 1755-1757.
- Task Force on Genetic Testing. Promoting Safe and Effective Genetic Testing in the United States: Final Report.1997 (in press)
- Rothenberg KH.Genetic information and health insurance: State legislative approaches. J Law Med Ethics 1995; 23: 312-319.
- Alper JS, Geller LN, Barash CI et al. Genetic discrimination and screening for hemochromatosis. J Public Health Policy.1994; 15: 345-358.
- Haynes RB, Sackett DL, Taylor DW, Gibson ES, Johnson AL. Increased absenteeism after detection and labelling of hypertensive patients. N Engl J Med 1978; 299: 741-744.
- Brett AS. Psychologic effects of the diagnosis and treatment of hypercholesterolemia: lessons from case studies. Am J Med 1992; 91: 842-847.
- Irvine MJ, Logan AG. Is knowing your cholesterol number harmful? J Clin Epidemiol 1994; 47: 131-145.
- Marteau TM, Kinmouth AL, Thompson S, Pyke S. The psychological impact of cardiovascular screening and intervention in primary care: a problem of false reassurance? Br J Gen Practice.1996; 46: 577-582.
- Markel H. The stigma of disease: implications of genetic screening. Am J Med 1992; 93: 209-215.
- Millar MG, Millar K. Negative affective consequences of thinking about disease detection behaviors. Health Psychol 1995; 14: 141-146.
- Sempos CT, Cleeman JI, Carroll MD et al. Prevalence of high blood cholesterol among US adults. JAMA 1993; 269: 3009-3014.
- Page last reviewed: January 1, 2009 (archived document)
- Content source: