Tumor hypoxia
Tumor hypoxia is the situation where tumor cells have been deprived of oxygen. As a tumor grows, it rapidly outgrows its blood supply, leaving portions of the tumor with regions where the oxygen concentration is significantly lower than in healthy tissues. Hypoxic microenvironements in solid tumors are a result of available oxygen being consumed within 70 to 150 μm of tumour vasculature by rapidly proliferating tumor cells thus limiting the amount of oxygen available to diffuse further into the tumor tissue. In order to support continuous growth and proliferation in challenging hypoxic environments, cancer cells are found to alter their metabolism. Furthermore, hypoxia is known to change cell behavior and is associated with extracellular matrix remodeling and increased migratory and metastatic behavior.[1][2]
Changes in the glycolytic pathway
A particular change in metabolism, historically known as the Warburg effect[3] results in high rates of glycolysis in both normoxic and hypoxic cancer cells. Expression of genes responsible for glycolytic enzymes and glucose transporters are enhanced by numerous oncogenes including RAS, SRC, and MYC.[4][5]
HIF-1 induced changes in gene expression
Traditionally, hypoxia leads to increased production of hypoxia-inducible factor (HIF-1), containing HIF-1α and HIF-1β subunits, that acts as a key regulatory transcription factor responsible for adaptive cellular changes. In humans, HIF-1 has been shown to up-regulate expression of genes affecting a range of target areas of physiology. These genes range from those involved in triggering an inflammatory response to those responsible for iron metabolism. Particularly notable when focusing on metabolism, HIF-1 is shown to affect glycolytic genes to cope with reductions in oxygen availability and consumption.
These genes include: solute carrier family 2 (GLUT1), hexokinase (HK), phosphoglucose isomerase (PGI), phosphofructokinase (PFKL), fructose-bisphosphate aldolase (ALDO), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGM), enolase 1 (ENOA), pyruvate kinase (PK), pyruvate dehydrogenase kinase, isozyme 1 (PDK1) and lactate dehydrogenase A (LDH-A).[6]
In addition to alterations in oxygen concentration associated with hypoxic microenvironments, glucose concentration gradients found in tumors also influence the rate of aerobic and anaerobic glycolysis. A carbohydrate-response element (ChoRE) is responsible for regulating glycolytic enzyme gene expression in response to changing glucose concentrations through a binding interaction at the same consensus sequence as HIF-1. Interactions of HIF-1 and ChoRE with the DNA sequence 5’-RCGTG-3’ leads to increased expression of genes listed above.[7]
GLUT1 transporter expression
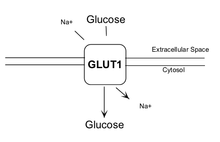
GLUT1 is a member of the GLUT transporter family of 14 hexose transporters responsible for facilitating the transport of hexose sugars along the concentration gradient. GLUT1 is the most abundantly expressed of the family thought to maintain basal glucose transport in almost all cell types. GLUT1 levels, in response to hypoxic conditions, have been shown to increase with changes at both the mRNA and protein levels.[8] Moreover, transport of GLUT1 has been shown to increase under these hypoxic conditions. With the role of transporting sugars from the extracellular to the intracellular environment, GLUT1, along with other members of the GLUT family, can be rate-controlling for cellular glycolytic metabolism. Having an increased level of GLUT1, in the case of hypoxic tumors, increases the flux of glucose into the cells allowing for a higher rate of glycolysis and thus greater risks of metastasis (as elaborated upon below).[9]
Hexokinase 2 expression
Hexokinase (HK) is the first enzyme in the glycolytic pathway converting glucose to glucose-6-phosphate through an ATP-dependent phosphorylation event. Important for glycolysis to proceed, the hexokinase reaction activates glucose for subsequent steps. In hypoxic tumors, hexokinase mRNA abundance is significantly increased as well as protein levels.[10] Increased expression of hexokinase 2, in some cases nearly 10-fold, allows for an increased flux of glucose through the glycolytic pathway subsequent to the increased uptake by GLUT1.<[11]
| D-Glucose | Hexokinase Expression up-regulated by HIF-1 | α-D-Glucose 6-phosphate | |
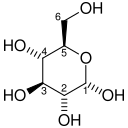 |
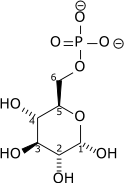 | ||
| ATP | ADP | ||
| PO3- 4 |
H2O | ||
| Glucose 6-phosphatase | |||
Phosphoglucose isomerase expression
Phosphoglucose isomerase (PGI) is a housekeeping cytosolic enzyme with roles in both glycolysis and gluconeogenesis pathways. It is responsible for catalyzing the interconversion of glucose 6-phosphate and fructose 6-phosphate. Extracellularly, PGI is known as an autocrine motility factor (AMF) eliciting mitogenic, motogenic, differentiation functions as well as tumor progression and metastasis.[12] Activation of PGI through proposed HIF-1 induced mechanisms results in increased conversion of glucose 6-phosphate to fructose 6-phosphate and also contributes to cell motility and invasion during cancer metastasis.
| α-D-Glucose 6-phosphate | Phosphoglucose isomerase Expression up-regulated by HIF-1 | β-D-Fructose 6-phosphate | |
|
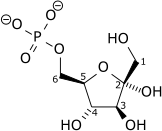 | ||
| Phosphoglucose isomerase | |||
6-Phosphofructo-2-kinase/fructose 2,6-bisphosphatases expression
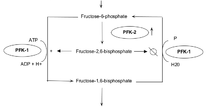
6-Phosphofructo-2-kinases/fructose 2,6-bisphosphatases (PFKFBs) belong to a family of bifunctional ATP-dependent enzymes responsible for controlling the level of glycolysis intermediate fructose-1,6-bisphosphate. HIF-1-induced expression of these enzymes (PFK-2/FBPase-2) subsequently alters the balance of fructose-2,6-bisphosphate which plays an important role as an allosteric activator of phospho-fructokinase 1 (PFK-1). PFK-1 is an enzyme that controls one of the most critical steps of glycolysis. Regulation of PFK-1 is also mediated by the cellular energy status in result of ATP's inhibitory effect. Greater quantities of fructose-2,6-bisphosphate in cancer cells, in result of HIF-1 expression of PFK-2/FBPase-2, thus activates PFK-1allowing for an increased glycolytic flux converting fructose-6-phosphate to fructose-1,6-bisphosphate. Allosteric regulation of glycolysis by fructose-2,6-bisphosphate allows cancer cells to maintain a glycolytic balance to match their bioenergetic and biosynthetic demands.[13]
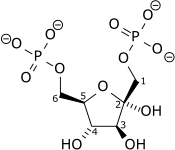
| β-D-Fructose 6-phosphate (F6P) | phosphofructokinase (PFK-1) Expression up-regulated by HIF-1 |
β-D-Fructose 1,6-bisphosphate (F1,6BP) | |
|
 | ||
| ATP | H+ + ADP | ||
Fructose-1,6-bisphosphate aldolase expression
Fructose-1,6-bisphosphate aldolase (ALDO) belongs to a family include aldolase A, B and C. Unique in glycolysis, aldolase enzymes cleave fructose-1,6-bisphosphate into two 3-C molecules including glyceraldehyde-3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP). With the HIF-1 mediated expression of aldolase A under hypoxic conditions, the catalysis of fructose-2,6-bisphosphate to glyceraldehyde-3-phosphate and dihydroxyacetone phosphate is increased thus leading to increased glycolytic flux.[14]
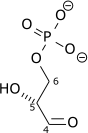
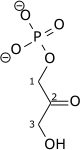
| β-D-Fructose 1,6-bisphosphate (F1,6BP) | fructose-1,6-bisphosphate aldolase Expression up-regulated by HIF-1 |
D-glyceraldehyde 3-phosphate (GADP) | Dihydroxyacetone phosphate (DHAP) | ||
|
 |
+ |  | ||
Glyceraldehyde-3-phosphate dehydrogenase expression
The glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), is responsible for the oxidative conversion of glyceraldehyde-3-phosphate (GADP) to 1,3-bisphosphoglycerate (1,3BPG). Up-regulation of glyceraldehyde-3-phosphate dehydrogenase expression is maximal (4-5 fold) following hypoxic conditions of ~24 hours in vascular endothelial cells.[15] Various models for the exact glyceraldehyde-3-phosphate dehydrogenase activation mechanisms have been proposed.
| glyceraldehyde 3-phosphate (GADP) | glyceraldehyde phosphate dehydrogenase Expression up-regulated by HIF-1 |
D-1,3-bisphosphoglycerate (1,3BPG) | |
|
 | ||
| NAD+ + Pi | NADH + H+ | ||
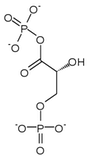
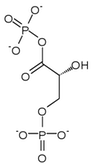
Phosphoglycerate kinase 1 expression
Hypoxia has been shown to induce a 10-fold accumulation of phosphoglycerate kinase 1 (PGK-1) mRNA in mouse hepatoma (Hepa 1c1c7) cells. Phosphoglycerate kinase 1 is an enzyme involved in the conversion of 1,3-bisphosphoglycerate (1,3-BPG) to 3-phosphoglycerate (3-P-G) leading the production of ATP from ADP. Induction of gene expression by HIF-1 is thought to be dependent on the presence of aromatic hydrocarbon receptor nuclear translocator (ARNT1). Arnt's N-terminal region and HIF-1 are thought to work together to induce transcription of phosphoglycerate kinase 1.[16]
| 1,3-bisphosphoglycerate (1,3-BPG) | phosphoglycerate kinase Expression up-regulated by HIF-1 |
3-phosphoglycerate (3-P-G) | |
 |
 | ||
| ADP | ATP | ||
| phosphoglycerate kinase | |||
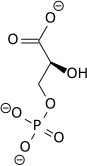
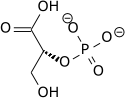
Phosphoglycerate mutase expression
Phosphoglycerate mutase B (PGM-B) is one of the latter glycolytic enzymes responsible for the conversion of 3-phosphoglycerate (3PG) to 2-phosphoglycerate (2PG). Both protein and mRNA levels were shown to increase 2-3-fold in research exposing fetal rat lung fibroblasts to hypoxic conditions. Increased levels appeared to be regulated at the transcriptional level as per many of the other glycolytic enzymes. Maximum up regulation was shown following 16 hours thus supporting its role in contributing to an increased glycolytic flux for adaption of cells to hypoxia.[17]
| 3-phosphoglycerate (3PG) | phosphoglycerate mutase Expression up-regulated by HIF-1 |
2-phosphoglycerate (2PG) | |
|
 | ||
Enolase 1 expression
Enolase 1, also known as α-enolase, is encoded by the ENOA gene and is responsible for converting 2-phosphoglycerate to phosphoenolpyruvate in the glycolytic pathway. Both enolase 1 overexpression and its post-translational modifications could be of value for diagnostic and prognostic work in terms of cancer. Although the exact roles of post-translational modifications have not been completely elucidated, patterns are shown between certain cancer cell types suggesting they may have important influence on function, localization and immunogenicity.[18] Aside from its role in promoting glycolytic flux and anearobic energy production, it has been shown to induce a specific humoral and cellular immune response. On all levels, hypoxia-induced over-expression of enolase 1 may possess important roles in hypoxic tumors including the most straightforward increase in anearobic respiration.
| 2-phosphoglycerate (2PG) | enolase 1 Expression up-regulated by HIF-1 |
phosphoenolpyruvate (PEP) | |
|
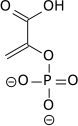 | ||
| H2O | |||
| enolase 1 | |||
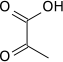
Pyruvate kinase expression
HIF-1 activated pyruvate kinase M comes in multiple isoforms known as PKM1 and PKM2. Pyruvate kinase is shown to convert phosphoenolpyruvate to pyruvate forming ATP from ADP. Along with phospho-fructokinase 1, pyruvate kinase is also allosterically activated by fructose-2,6-bisphosphate. In cancer cells pyruvate kinase M2 has been shown to interact directly with HIF-1α enhancing HIF-1 binding and p300 recruitment to hypoxia response elements. This positive feedback loop leads to HIF-1 transactivation and an amplified effect on glucose metabolism.[19]
Pyruvate kinase M2 is often considered the main regulator of cancer metabolism with roles in various parallel, feed-forward, positive and negative feedback mechanisms. The genetic difference between pyruvate kinase M1 and pyruvate kinase M2 is only 22 out of 531 amino acids which makes an immense difference. Pyruvate kinase M2 has metabolic activity regulated by post-translational modifications including acetylation, oxidation, phosphorylation, hydroxylation and sumoylation. These different modifications can cause the shift from the metabolically active tetrameric form to the in-active monomeric form. The well-known EGFR-activated extracellular signal-regulated kinase 2 (ERK2) and death-associated protein kinase are both shown to bind and directly phosphorylate pyruvate kinase M2 leading to increased activity in the glycolysis pathway.[20] In hypoxic conditions found in a solid tumor, pyruvate kinase M2 plays a large role in promoting anearobic energy production.
| phosphoenolpyruvate (PEP) | pyruvate kinase Expression up-regulated by HIF-1 |
pyruvate (Pyr) | |
|
 | ||
| ADP + H+ | ATP | ||
Pyruvate dehydrogenase kinase expression
Pyruvate dehydrogenase directly follows the glycolytic pathway and is responsible for the conversion of pyruvate to acetyl-CoA which enters into the TCA cycle. The TCA cycle, although not directly requiring oxygen, requires the cycling of NADH to NAD+ as performed by the electron transport chain under aerobic conditions. Under anaerobic conditions, such as those found in hypoxic tumors, the TCA cycle provides little ATP yield due to the lack of electron transport chain function. In order to direct the glycolytically produced pyruvate away from the TCA cycle, pyruvate dehydrogenase kinase is over-expressed in response to hypoxic conditions. Pyruvate dehydrogenase kinase is not a glycolytic enzymes but more of a glycolytic regulator. Pyruvate dehydrogenase kinases, transcriptionally activated by HIF-1 in hypoxic conditions, are responsible for phosphorylating the E1 subunit of pyruvate dehydrogenase ultimately suppressing its function.[21] By inhibiting this specific pathway, the glycolytic products are directed away from the mitochondrial TCA cycle and towards lactate dehydrogenase.[22]
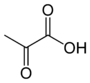
Lactate dehydrogenase expression
Activated expression of lactate dehydrogenase A (LDH-A), parallels with deactivation of pyruvate dehydrogenase mediated by pyruvate dehydrogenase kinase. Subsequent inactivation of pyruvate dehydrogenase following phosphorylation and increased expression of lactate dehydrogenase A shunts pyruvate away from the mitochondrial TCA cycle. In many different tumor types lactate dehydrogenase A is found at elevated levels and has even been linked to poor prognosis and a greater metastatic potential[23] The high levels of lactate production surface the question of whether lactate has some influence on the aggressive behaviour shown in hypoxic tumors.
| pyruvate | lactate dehydrogenase Expression up-regulated by HIF-1 |
Lactate | |
 |
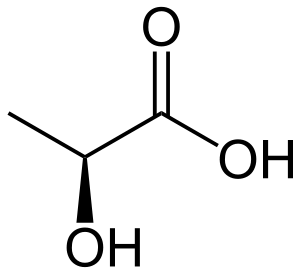 | ||
| NADH | NAD+ | ||
| lactate dehydrogenase | |||
Overview of glycolytic changes and consequences
Increased expression of almost every glycolytic enzyme is seen in hypoxic tumor conditions. The over-expression of these proteins is mediated by HIF-1 and completely alters normal cellular metabolism. With decreases in the rate of mitochondrial oxidation, lactate and protons begin to accumulate. High levels of glycolysis and the production of lactate, as shown in hypoxic tumor cells, is hallmark of cancer cells even in the presence of oxygen.
To relieve tumors cells of acidosis, carbonic anhydrases appear to be highly expressed once again downstream of HIF-1 activation. These enzymes catalyze the reversible hydration of carbon dioxide into bicarbonate and protons. They also assist in acidifying the extracellular environment and maintaining a slightly alkaline intracellular compartments contributing to tumor cell survival.[24] Lactate from the hypoxic tumor cells is excreted to the surrounding environment by carbonic anhydrase 9 and sodium-hydrogen exchanger 1 MCT4. Local aerobic cancer cells are though to take up this lactate forming a metabolic symbiosis.[25]
Lactate and cancer
It is commonly accepted that cancer cells (both hypoxic and normoxic) produce large amounts of lactate in result of a large metabolic shift from oxidative phosphorylation to altered glycolysis. The high levels of released lactate contribute to immune escape for the tumor cells. Activated T cells use glycolysis as an energy source and thus must regulate their own lactate levels. Traditionally done by a secretion method, immune cells in a lactate rich environment cannot rid themselves of their own lactate due to the concentration gradient. It is thought that leukocytes may be asphyxiated by lactate while low extracellular pHs may also reduce cytotoxic T-cell function.[26]
In endothelial cells it has also been shown that lactate stimulates vascular endothelial growth factor (VEGF) production leading to enhanced cellular migration in result of lactate-induced angiogenesis[27] Recent work has also uncovered that lactate uptake by MCT-1 in endothelial cells stimulates NF-κB activation and thus IL-8 expression. Release of lactate from tumor cells through MCT-4 was sufficient to stimulate angiogenesis and tumor growth through an IL-8-dependent mechanism.
Lactate has demonstrated the ability to increase hyaluronan production leading to elevated expression of CD44. Hyaluronan is a glycosaminoglycan polymer critical for maintaining extracellular matrix integrity and modulating cell-cell interactions. Hyaluronan is tethered to cell surfaces by CD44 which are anchored in caveolin-rich lipid rafts. Cleavage and further degradation of hyaluronan is facilitated by Hyal2 and Hyal1, respectively.[28] Increased levels of hyaluronan surrounding carcinomas leads to the promotion of cellular growth and motility. A lactate-sensitive response element for genes in fibroblasts involved in hyaluronan metabolism has been identified.
Finally, it is also worth noting that lactate concentrations are positively correlated with radioresistance. Many anticancer therapies, including ionizing radiation and many chemotherapeutics, rely on the overproduction of reactive oxygen species to cause genomic instability. Lactate, as an antioxidant, may act to scrub down the levels of reactive oxygen species thus enhancing resistance to radiation and chemotherapy.[29]
Acidic microenvironment and metastasis
It is thought that the low pH of hypoxic tumors in result of high levels of lactic acid can promote tumor cell invasion by destruction of adjacent non-cancerous tissue.[30]> Carbonic anhydrase 9 involved in maintaining a slightly alkaline intracellular pH does so by removing carbonate from the extracellular space consequently acidifying the cells surroundings. Moreover, proton pumping from the hypoxic tumor cells further decreases the surrounding pH. On a completely different note, as briefly discussed above, the autocrine function of phosphoglucose isomerase also promotes cell motility and metastasis.
Metabolic symbiosis
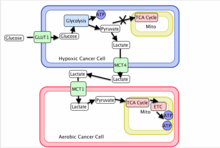
With the hypoxic tumor cells consuming large amounts of glucose to maintain energy homeostasis, the tumor has found a way of using its resources most efficiently. The end glycolytic product of hypoxic tumors, lactate, is transported out of the hypoxic cell by monocarboxylate transporter 4 (MCT4) which is a hypoxia induced transporter. The free lactate in the extracellular space is then taken up by monocarboxylate transporter 1 (MCT1) which is a non-hypoxia induced transporter found on the surface of aerobic cells. This transporter allows aerobic cancer cells to efficiently take up lactate, convert it back to pyruvate with the oxygen-dependent expression of lactate dehydrogenase B (LDH-B), and use it as an energy source. This frees these cells from requiring large quantities of glucose allowing the hypoxic cells to take up the majority of the available resources.
Tumor cells have also shown the remarkable ability to adapt to regional variation of oxygen availability. Cancer cells demonstrate the ability to be hypoxic at one point in time and aerobic at the next.[31] This shows cyclic variations in oxygenation implying dynamic regulation of the metabolic symbiosis between lactate-producing and lactate-consuming states.
The pentose phosphate pathway
In order to meet the demands of rapid tumor growth, the tumor must find ways to support the synthesis of a complete daughter cell while facing depleting nutrient supplies. They must coordinate production of precursors for macromolecular synthesis as well as maintain cellular bioenergetics without impairing cell growth, proliferation and viability. One way of doing this is by shuffling glycolytic intermediates such as glucose-6-phosphate into the pentose phosphate pathway to give ribose-5-phosphate and NADPH. Ribose-5-phosphate acts as an intermediate for the production of nucleotides thus providing a connection between glycolysis and nucleotide synthesis in hypoxic tumor cells. In cases where glycolysis remains highly active in normoxic conditions, NADPH acts as a mediator of antioxidative reactions to protect cells from oxidative damage.[32]
Cancer treatments and tumor hypoxia
Radiotherapy
The presence or absence of oxygen has a strong influence on ionizing radiation to cause cell death of tumour and normal cells.[33] This is called the oxygen effect. Under hypoxic conditions it has been shown that cells obtain radioresistance through HIF-1 mediated mechanisms. To overcome this problem, radiation oncologists have developed powerful tools and approaches such as simultaneous integrated boost intensity-modulated radiation therapy (SIB-IMRT), which enables a booster dose of radiation to be delivered to small target fractions in a malignant tumor, hypoxia-selective cytotoxins/drugs, and HIF-1 inhibitors.[34] Moreover, it is possible to treat a hypoxic tumor by means of ion beam therapy, in particular with 12C. As ions' damage is direct, OER (Oxygen Enhancement Ratio) is 1, so the effect of oxygen it is not important.
Other treatment options
Bioreductive prodrugs play a significant part in dealing with these kinds of cells: they can kill the oxygen-deficient tumor cells selectively as hypoxia-activated prodrugs. Example drugs include Tirapazamine and Evofosfamide. The study of tumors in such conditions was pioneered by Dr L. H. Gray.
Targeting tumor hypoxia to overcome metastasis
An association between tumor hypoxia and metastatic progression has been shown through numerous publications.[35][36]
Drug development
Several approaches have been taken to address tumor hypoxia. Some companies tried to develop drugs that are activated in hypoxic environments (Novacea, Inc. Proacta, Inc, and Threshold Pharmaceuticals, Inc), while others are currently seeking to reduce tumor hypoxia (Diffusion Pharmaceuticals, Inc. and NuvOx Pharma, LLC).
Several companies have tried to develop drugs that are activated in hypoxic environments. These drug candidates target levels of hypoxia that are common in tumors but are rare in normal tissues. The hypoxic zones of tumors generally evade traditional chemotherapeutic agents and ultimately contribute to relapse. In the literature, hypoxia has been demonstrated to be associated with a worse prognosis, making it a determinant of cancer progression and therapeutic response[35] Several review articles summarize the current status of hypoxic cytotoxins (hypoxia activated prodrugs).[37][38][39] Companies that have tried drugs that are activated in hypoxic environments included Novacea, Inc. Proacta, and Threshold Pharmaceuticals. Novacea Inc discontinued development of its hypoxia activated drug.[40] Proacta's drug PR610 failed a Phase I clinical trial due to toxicity.[41] Threshold Pharmaceuticals discontinued the hypoxia activated prodrug, TH-302, after Phase III trials failed to show statistically significant overall survival.[42]
Niacinamide, the active form of vitamin B3, acts as a chemo- and radio-sensitizing agent by enhancing tumor blood flow, thereby reducing tumor hypoxia. Niacinamide also inhibits poly(ADP-ribose) polymerases (PARP-1), enzymes involved in the rejoining of DNA strand breaks induced by radiation or chemotherapy.[43] As of August 2016, no clinical trials appear to be in progress for this indication.
Another approach to the treatment of tumor hypoxia is the use of an oxygen diffusion-enhancing compound to reoxygenate the hypoxic zones of tumors. The developer of oxygen diffusion-enhancing compounds, Diffusion Pharmaceuticals, tested its lead compound, trans sodium crocetinate (TSC), in a Phase II clinical trial in 59 patients newly diagnosed with glioblastoma multiforme.[44] The results of the Phase II showed that 36% of the full-dose TSC patients were alive at 2 years, compared with historical survival values ranging from 27% to 30% for the standard of care.[45] The main endpoint of the trial was survival at two years, not overall survival.[44]
Another drug in development that is designed to reduce tumor hypoxia is NuvOx Pharma's NVX-108. NVX-108 is a formulation of the perfluorocarbon, dodecafluoropentane (DDFPe). NVX-108 is injected intravenously, flows through the lungs and picks up oxygen, then flows through the arteries and releases oxygen in the presence of hypoxic tissue. A Phase Ib/II clinical trial is in progress for newly diagnosed glioblastoma multiforme.[46] Early results have shown reversal of tumor hypoxia, and the trial continues to progress.[47]
See also
References
- Gilkes DM, Semenza GL, Wirtz D (June 2014). "Hypoxia and the extracellular matrix: drivers of tumour metastasis". Nature Reviews. Cancer. 14 (6): 430–9. doi:10.1038/nrc3726. PMC 4283800. PMID 24827502.
- Spill F, Reynolds DS, Kamm RD, Zaman MH (August 2016). "Impact of the physical microenvironment on tumor progression and metastasis". Current Opinion in Biotechnology. 40: 41–48. doi:10.1016/j.copbio.2016.02.007. PMC 4975620. PMID 26938687.
- Vander Heiden MG, Cantley LC, Thompson CB (May 2009). "Understanding the Warburg effect: the metabolic requirements of cell proliferation". Science. 324 (5930): 1029–33. Bibcode:2009Sci...324.1029V. doi:10.1126/science.1160809. PMC 2849637. PMID 19460998.
- Flier JS, Mueckler MM, Usher P, Lodish HF (March 1987). "Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes". Science. 235 (4795): 1492–5. Bibcode:1987Sci...235.1492F. doi:10.1126/science.3103217. PMID 3103217.
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. (July 2000). "Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc". The Journal of Biological Chemistry. 275 (29): 21797–800. doi:10.1074/jbc.C000023200. PMID 10823814.
- Kanehisa M (2013). "Molecular network analysis of diseases and drugs in KEGG". Methods in Molecular Biology. 939: 263–75. doi:10.1007/978-1-62703-107-3_17. ISBN 978-1-62703-106-6. PMID 23192552.
- Dang CV, Semenza GL (February 1999). "Oncogenic alterations of metabolism". Trends in Biochemical Sciences. 24 (2): 68–72. doi:10.1016/S0968-0004(98)01344-9. PMID 10098401.
- Zhang JZ, Behrooz A, Ismail-Beigi F (July 1999). "Regulation of glucose transport by hypoxia". American Journal of Kidney Diseases. 34 (1): 189–202. doi:10.1016/s0272-6386(99)70131-9. PMID 10401038.
- Airley R, Loncaster J, Davidson S, Bromley M, Roberts S, Patterson A, et al. (April 2001). "Glucose transporter glut-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix". Clinical Cancer Research. 7 (4): 928–34. PMID 11309343.
- Yasuda, Seiichi, et al. "Hexokinase II and VEGF expression in liver tumors: correlation with hypoxia-inducible factor-1α and its significance." Journal of Hepatology 40.1 (2004): 117-123.
- Natsuizaka M, Ozasa M, Darmanin S, Miyamoto M, Kondo S, Kamada S, et al. (September 2007). "Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia". Experimental Cell Research. 313 (15): 3337–48. doi:10.1016/j.yexcr.2007.06.013. hdl:2115/29921. PMID 17651733.
- Funasaka T, Yanagawa T, Hogan V, Raz A (September 2005). "Regulation of phosphoglucose isomerase/autocrine motility factor expression by hypoxia". FASEB Journal. 19 (11): 1422–30. doi:10.1096/fj.05-3699com. PMID 16126909.
- Ros S, Schulze A (February 2013). "Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism". Cancer & Metabolism. 1 (1): 8. doi:10.1186/2049-3002-1-8. PMC 4178209. PMID 24280138.
- Lorentzen E, Siebers B, Hensel R, Pohl E (March 2005). "Mechanism of the Schiff base forming fructose-1,6-bisphosphate aldolase: structural analysis of reaction intermediates". Biochemistry. 44 (11): 4222–9. doi:10.1021/bi048192o. PMID 15766250.
- Graven KK, McDonald RJ, Farber HW (February 1998). "Hypoxic regulation of endothelial glyceraldehyde-3-phosphate dehydrogenase". The American Journal of Physiology. 274 (2): C347–55. doi:10.1152/ajpcell.1998.274.2.C347. PMID 9486123.
- Li H, Ko HP, Whitlock JP (August 1996). "Induction of phosphoglycerate kinase 1 gene expression by hypoxia. Roles of Arnt and HIF1alpha". The Journal of Biological Chemistry. 271 (35): 21262–7. doi:10.1074/jbc.271.35.21262. PMID 8702901.
- Takahashi Y, Takahashi S, Yoshimi T, Miura T (June 1998). "Hypoxia-induced expression of phosphoglycerate mutase B in fibroblasts". European Journal of Biochemistry. 254 (3): 497–504. doi:10.1046/j.1432-1327.1998.2540497.x. PMID 9688259.
- Capello M, Ferri-Borgogno S, Cappello P, Novelli F (April 2011). "α-Enolase: a promising therapeutic and diagnostic tumor target". The FEBS Journal. 278 (7): 1064–74. doi:10.1111/j.1742-4658.2011.08025.x. PMID 21261815.
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. (May 2011). "Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1". Cell. 145 (5): 732–44. doi:10.1016/j.cell.2011.03.054. PMC 3130564. PMID 21620138.
- Filipp FV (2013). "Cancer metabolism meets systems biology: Pyruvate kinase isoform PKM2 is a metabolic master regulator". Journal of Carcinogenesis. 12: 14. doi:10.4103/1477-3163.115423. PMC 3746496. PMID 23961261.
- Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL (January 2005). "Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma". Neoplasia. 7 (1): 1–6. doi:10.1593/neo.04373. PMC 1490315. PMID 15736311.
- Kim JW, Dang CV (September 2006). "Cancer's molecular sweet tooth and the Warburg effect". Cancer Research. 66 (18): 8927–30. doi:10.1158/0008-5472.CAN-06-1501. PMID 16982728.
- Serganova I, Rizwan A, Ni X, Thakur SB, Vider J, Russell J, et al. (October 2011). "Metabolic imaging: a link between lactate dehydrogenase A, lactate, and tumor phenotype". Clinical Cancer Research. 17 (19): 6250–6261. doi:10.1158/1078-0432.CCR-11-0397. PMC 4217119. PMID 21844011.
- Chiche J, Ilc K, Laferrière J, Trottier E, Dayan F, Mazure NM, et al. (January 2009). "Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH". Cancer Research. 69 (1): 358–68. doi:10.1158/0008-5472.CAN-08-2470. PMID 19118021.
- Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. (December 2008). "Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice". The Journal of Clinical Investigation. 118 (12): 3930–42. doi:10.1172/JCI36843. PMC 2582933. PMID 19033663.
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. (May 2007). "Inhibitory effect of tumor cell-derived lactic acid on human T cells". Blood. 109 (9): 3812–9. doi:10.1182/blood-2006-07-035972. PMID 17255361.
- Beckert S, Farrahi F, Aslam RS, Scheuenstuhl H, Königsrainer A, Hussain MZ, Hunt TK (2006). "Lactate stimulates endothelial cell migration". Wound Repair and Regeneration. 14 (3): 321–4. doi:10.1111/j.1743-6109.2006.00127.x. PMID 16808811.
- Stern R (August 2008). "Hyaluronidases in cancer biology". Seminars in Cancer Biology. 18 (4): 275–80. doi:10.1016/j.semcancer.2008.03.017. PMID 18485730.
- Sattler UG, Mueller-Klieser W (November 2009). "The anti-oxidant capacity of tumour glycolysis". International Journal of Radiation Biology. 85 (11): 963–71. doi:10.3109/09553000903258889. PMID 19895273.
- Gort EH, Groot AJ, van der Wall E, van Diest PJ, Vooijs MA (February 2008). "Hypoxic regulation of metastasis via hypoxia-inducible factors". Current Molecular Medicine. 8 (1): 60–7. doi:10.2174/156652408783565568. PMID 18289014.
- Cárdenas-Navia LI, Mace D, Richardson RA, Wilson DF, Shan S, Dewhirst MW (July 2008). "The pervasive presence of fluctuating oxygenation in tumors". Cancer Research. 68 (14): 5812–9. doi:10.1158/0008-5472.CAN-07-6387. PMID 18632635.
- DeBerardinis RJ (November 2008). "Is cancer a disease of abnormal cellular metabolism? New angles on an old idea". Genetics in Medicine. 10 (11): 767–77. doi:10.1097/GIM.0b013e31818b0d9b. PMC 2782690. PMID 18941420.
- Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC (December 1953). "The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy". The British Journal of Radiology. 26 (312): 638–48. doi:10.1259/0007-1285-26-312-638. PMID 13106296.
- Harada H (2011). "How can we overcome tumor hypoxia in radiation therapy?". Journal of Radiation Research. 52 (5): 545–56. Bibcode:2011JRadR..52..545H. doi:10.1269/jrr.11056. PMID 21952313.
- Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P (October 1996). "Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix". Cancer Research. 56 (19): 4509–15. PMID 8813149.
- Vergis R, Corbishley CM, Norman AR, Bartlett J, Jhavar S, Borre M, et al. (April 2008). "Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study". The Lancet. Oncology. 9 (4): 342–51. doi:10.1016/S1470-2045(08)70076-7. PMID 18343725.
- Brown JM, Wilson WR (June 2004). "Exploiting tumour hypoxia in cancer treatment". Nature Reviews. Cancer. 4 (6): 437–47. doi:10.1038/nrc1367. PMID 15170446.
- Ahn GO, Brown M (May 2007). "Targeting tumors with hypoxia-activated cytotoxins". Frontiers in Bioscience. 12 (8–12): 3483–501. doi:10.2741/2329. PMID 17485316.
- McKeown SR, Cowen RL, Williams KJ (August 2007). "Bioreductive drugs: from concept to clinic". Clinical Oncology. 19 (6): 427–42. doi:10.1016/j.clon.2007.03.006. PMID 17482438.
- "Transcept Pharmaceuticals to merge with Novacea, move sleep drug forward". bizjournals.com. August 2016.
- "A Dose Escalation Trial of PR610 Treating Patients With Solid Tumors". ClinicalTrials.gov. August 2016.
- "Threshold Pharmaceuticals Announces Workforce Reduction". Fierce Biotech. December 2015.
- "Definition of niacinamide - National Cancer Institute Drug Dictionary - National Cancer Institute". Cancer.gov. 2011-02-02. Retrieved 2011-12-21.
- "Safety and Efficacy Study of Trans Sodium Crocetinate (TSC) With Concomitant Radiation Therapy and Temozolomide in Newly Diagnosed Glioblastoma (GBM)". ClinicalTrials.gov. November 2011.
- Gainer JL, Sheehan JP, Larner JM, Jones DR (February 2017). "Trans sodium crocetinate with temozolomide and radiation therapy for glioblastoma multiforme". Journal of Neurosurgery. 126 (2): 460–466. doi:10.3171/2016.3.JNS152693. PMID 27177177.
- "The Effects of NVX-108 as a Radiation Sensitizer in Glioblastoma (GBM))". ClinicalTrials.gov. August 2016.
- "NuvOx Reports Favorable Data on Phase Ib Clinical Trial in Glioblastoma". AZBio. July 2015.
Further reading
- Sullivan R, Graham CH (June 2007). "Hypoxia-driven selection of the metastatic phenotype". Cancer Metastasis Reviews. 26 (2): 319–31. doi:10.1007/s10555-007-9062-2. PMID 17458507.