Pathophysiology of Parkinson's disease
The pathophysiology of Parkinson's disease is death of dopaminergic neurons as a result of changes in biological activity in the brain with respect to Parkinson's disease (PD). There are several proposed mechanisms for neuronal death in PD; however, not all of them are well understood. Five proposed major mechanisms for neuronal death in Parkinson's Disease include protein aggregation in Lewy bodies, disruption of autophagy, changes in cell metabolism or mitochondrial function, neuroinflammation, and blood-brain barrier (BBB) breakdown resulting in vascular leakiness.[1]
| Neuronal Death in the PD brain | |
|---|---|
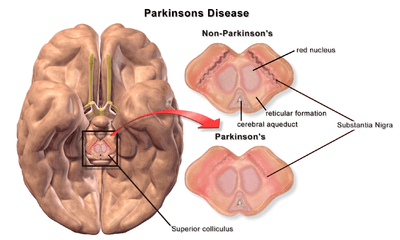 | |
| A brain without and with Parkinson's Disease compared in Substantia Nigra |
Protein aggregation
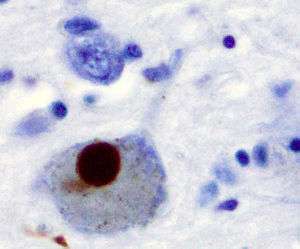
The first major proposed cause of neuronal death in Parkinson's disease is the bundling, or oligomerization, of proteins. The protein alpha-synuclein has increased presence in the brains of Parkinson's Disease patients and, as α-synuclein is insoluble, it aggregates to form Lewy bodies (shown to left) in neurons. Traditionally, Lewy bodies were thought to be the main cause of cell death in Parkinson's disease; however, more recent studies suggest that Lewy bodies lead to other effects that cause cell death.[2] Regardless, Lewy bodies are widely recognized as a pathological marker of Parkinson's disease.
Lewy bodies first appear in the olfactory bulb, medulla oblongata, and pontine tegmentum; patients at this stage are asymptomatic. As the disease progresses, Lewy bodies develop in the substantia nigra, areas of the midbrain and basal forebrain, and in the neocortex.
This mechanism is substantiated by the facts that α-synuclein lacks toxicity when unable to form aggregates; that heat-shock proteins, which assist in refolding proteins susceptible to aggregation, beneficially affect PD when overexpressed; and that reagents which neutralize aggregated species protect neurons in cellular models of α-synuclein overexpression.[3]
Alpha-synuclein appears to be a key link between reduced DNA repair and Parkinson’s disease.[4] Alpha-synuclein activates ATM (ataxia-telangiectasia mutated), a major DNA damage repair signaling kinase. Alpha-synuclein binds to breaks in double-stranded DNA and facilitates the DNA repair process of non-homologous end joining.[5] It was suggested [5] that cytoplasmic aggregation of alpha-synuclein to form Lewy bodies reduces its nuclear levels leading to decreased DNA repair, increased DNA double-strand breaks and increased programmed cell death of neurons.
Autophagy disruption
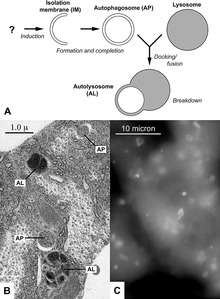
The second major proposed mechanism for neuronal death in Parkinson's disease, autophagy, is a mechanism by which inner components of the cell are broken down and recycled for use.[2][6] Autophagy has been shown to play a role in brain health, helping to regulate cellular function. Disruption of the autophagy mechanism can lead to several different types of diseases like Parkinson's disease.[6][7]
Autophagy dysfunction in Parkinson's disease has also been shown to lead to dysregulated mitochondria degradation.[8]
Changes in cell metabolism
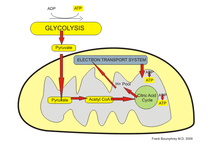
The third major proposed cause of cell death in Parkinson's disease involves the energy-generating mitochondrion organelle. In Parkinson's disease, mitochondrial function is disrupted, inhibiting energy production and resulting in death.[9][10]
The mechanism behind mitochondrial dysfunction in Parkinson's disease is hypothesized to be the PINK1 and Parkin complex, having been shown to drive autophagy of mitochondria (also known as mitophagy).[9][10][11] PINK1 is a protein normally transported into the mitochondrion, but can also accumulate on the surface of impaired mitochondria. Accumulated PINK1 then recruits Parkin; Parkin initiates the break down of dysfunctional mitochondria, a mechanism that acts as a "quality control"[9] In Parkinson's disease, the genes coding PINK1 and Parkin are thought to be mutated, therefore preventing the breakdown of impaired mitochondria, causing abnormal function and morphology of mitochondria and eventually cell death[9][10] Mitochondrial DNA (mtDNA) mutations have also been shown to accumulate with age[12] indicating that susceptibility to this mechanism of neuronal death increases with age.
Another mitochondrial-related mechanism for cell death in Parkinson's disease is the generation of reactive oxygen species (ROS).[12][13] ROS are highly reactive molecules that contain oxygen and can disrupt functions within the mitochondria and the rest of the cell. With increasing age, mitochondria lose their ability to remove ROS yet still maintain their production of ROS, causing an increase in net production of ROS and eventually cell death.[12][13]
As reviewed by Puspita et al.[14] studies have demonstrated that in the mitochondria and the endoplasmic reticulum, alpha-synuclein and dopamine levels are likely involved in contributing to oxidative stress as well as PD symptoms. Oxidative stress appears to have a role in mediating separate pathological events that together ultimately result in cell death in PD.[14] Oxidative stress leading to cell death may be the common denominator underlying multiple processes. Oxidative stress causes oxidative DNA damage. Such damage is increased in the mitochondria of the substantia nigra of PD patients and may lead to nigral neuronal cell death.[15][16]
Neuroinflammation
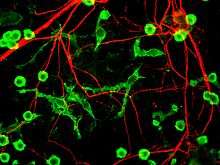
The fourth proposed major mechanism of neuronal death in Parkinson's Disease, neuroinflammation, is generally understood for neurodegenerative diseases, however, specific mechanisms are not completely characterized for PD.[17] One major cell type involved in neuroinflammation is the microglia. Microglia are recognized as the innate immune cells of the central nervous system. Microglia actively survey their environment and change their cell morphology significantly in response to neural injury. Acute inflammation in the brain is typically characterized by rapid activation of microglia. During this period, there is no peripheral immune response. Over time, however, chronic inflammation causes the degradation of tissue and of the blood–brain barrier. During this time, microglia generate reactive oxygen species and release signals to recruit peripheral immune cells for an inflammatory response.
In addition, microglia are known to have two major states: M1, a state in which cells are activated and secrete pro-inflammatory factors; and M2, a state in which cells are deactivated and secrete anti-inflammatory factors.[18] Microglia are usually in a resting state (M2), but in Parkinson's disease can enter M1 due to the presence of α-synuclein aggregates. The M1 microglia release pro-inflammatory factors which can cause motor neurons to die. In this case, dying cells can release factors to increase the activation of M1 microglia, leading to a positive feedback loop which causes continually increasing cell death.[17]
BBB breakdown
The fifth proposed major mechanism for cell death is the breakdown of the blood-brain barrier (BBB). The BBB has three cell types which tightly regulate the flow of molecules in and out of the brain: endothelial cells, pericytes, and astrocytes. In neurodegenerative diseases, BBB breakdown has been measured and identified in specific regions of the brain, including the substantia nigra in Parkinson's disease and hippocampus in Alzheimer's disease.[19] Protein aggregates or cytokines from neuroinflammation may interfere with cell receptors and alter their function in the BBB.[19][20] Most notably, vascular endothelial growth factor (VEGF) and VEGF receptors are thought to be dysregulated in neurodegenerative diseases. The interaction between the VEGF protein and its receptors leads to cell proliferation, but is believed to be disrupted in Parkinson's disease and Alzheimer's disease.[20][21] This then causes cells to stop growing and therefore, prevents new capillary formation via angiogenesis. Cell receptor disruption can also affect the ability for cells to adhere to one another with adherens junctions.[22]
Without new capillary formation, the existing capillaries break down and cells start to dissociate from each other. This in turn leads to the breakdown of gap junctions.[23][24] Gap junctions in endothelial cells in the BBB help prevent large or harmful molecules from entering the brain by regulating the flow of nutrients to the brain. However, as gap junctions break down, plasma proteins are able to enter in extracellular matrix the brain.[23] This mechanism is also known as vascular leakiness, where capillary degeneration leads to blood and blood proteins "leaking" into the brain. Vascular leakiness can eventually cause neurons to alter their function and shift towards apoptotic behavior or cell death.
Impact on locomotion

Dopaminergic neurons are the most abundant type of neuron in the substantia nigra, a part of the brain regulating motor control and learning. Dopamine is a neurotransmitter which activates motor neurons in the central nervous system. The activated motor neurons then transmit their signals, via action potential, to motor neurons in the legs.[25] However, when a significant percentage of the motor neurons die (about 50-60%), this decreases dopamine levels by up to 80%.[10] This inhibits the ability for neurons to generate and transmit a signal. This transmission inhibition ultimately causes the characteristic Parkinsonian gait with symptoms such as hunched and slowed walking or tremors.
References
- Tansey, M. G., & Goldberg, M. S. (2010). Neuroinflammation in Parkinson's disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease, 37(3), 510-518. doi:10.1016/j.nbd.2009.11.004
- Schapira, A. H. (2009). Etiology and Pathogenesis of Parkinson Disease. Neurologic Clinics, 27(3), 583-603. doi:10.1016/j.ncl.2009.04.004
- Stefanis, Leonidas (2012). "α-Synuclein in Parkinson's disease". Cold Spring Harb Perspect Med. 4.
- Abugable AA, Morris JLM, Palminha NM, Zaksauskaite R, Ray S, El-Khamisy SF. DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair (Amst). 2019 Sep;81:102669. doi: 10.1016/j.dnarep.2019.102669. Epub 2019 Jul 8. Review. PMID:31331820
- Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, Weston LJ, Owen N, Weissman TA, Luna E, Raber J, Luk KC, McCullough AK, Woltjer RL, Unni VK. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep. 2019 Jul 29;9(1):10919. PMID: 31358782 PMCID:PMC6662836 DOI: 10.1038/s41598-019-47227-z
- Stern, S. T., & Johnson, D. N. (2008). Role for nanomaterial-autophagy interaction in neurodegenerative disease. Autophagy, 4(8), 1097-1100. doi:10.4161/auto.7142
- Ghavami, S., Shojaei, S., Yeganeh, B., Ande, S. R., Jangamreddy, J. R., Mehrpour, M., . . . Łos, M. J. (2014). Autophagy and apoptosis dysfunction in neurodegenerative disorders. Progress in Neurobiology, 112, 24-49. doi:10.1016/j.pneurobio.2013.10.004
- Hu, Z., Yang, B., Mo, X., & Xiao, H. (2014). Mechanism and Regulation of Autophagy and Its Role in Neuronal Diseases. Molecular Neurobiology Mol Neurobiol, 52(3), 1190-1209. doi:10.1007/s12035-014-8921-4
- Chen, H., & Chan, D. C. (2009). Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Human Molecular Genetics, 18(R2). doi:10.1093/hmg/ddp326
- Pickrell, A., & Youle, R. (2015). The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron, 85(2), 257-273. doi:10.1016/j.neuron.2014.12.007
- Narendra, D., Tanaka, A., Suen, D., & Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol The Journal of Cell Biology, 183(5), 795-803. doi:10.1083/jcb.200809125
- Lin, M. T., & Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443(7113), 787-795. doi:10.1038/nature05292
- Jomova, K., Vondrakova, D., Lawson, M., & Valko, M. (2010). Metals, oxidative stress and neurodegenerative disorders. Molecular and Cellular Biochemistry Mol Cell Biochem, 345(1-2), 91-104. doi:10.1007/s11010-010-0563-x
- Puspita L, Chung SY, Shim JW (November 2017). "Oxidative stress and cellular pathologies in Parkinson's disease". Mol Brain. 10 (1): 53. doi:10.1186/s13041-017-0340-9. PMC 5706368. PMID 29183391.
- Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y (December 1999). "Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson's disease". Ann. Neurol. 46 (6): 920–4. PMID 10589547.
- Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K (April 2007). "Oxidative damage in nucleic acids and Parkinson's disease". J. Neurosci. Res. 85 (5): 919–34. doi:10.1002/jnr.21191. PMID 17279544.
- Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., & Gage, F. H. (2010). Mechanisms Underlying Inflammation in Neurodegeneration. Cell, 140(6), 918-934. doi:10.1016/j.cell.2010.02.016
- Zlokovic, B. V. (2008). The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron, 57(2), 178-201. doi:10.1016/j.neuron.2008.01.003
- Zlokovic, B. V. (2011). Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nature Reviews Neuroscience Nat Rev Neurosci. doi:10.1038/nrn3114
- Hao, T., & Rockwell, P. (2013). Signaling through the vascular endothelial growth factor receptor VEGFR-2 protects hippocampal neurons from mitochondrial dysfunction and oxidative stress. Free Radical Biology and Medicine, 63, 421-431. doi:10.1016/j.freeradbiomed.2013.05.036
- Almodovar, C. R., Lambrechts, D., Mazzone, M., & Carmeliet, P. (2009). Role and Therapeutic Potential of VEGF in the Nervous System. Physiological Reviews, 89(2), 607-648. doi:10.1152/physrev.00031.2008
- Förster, C., Burek, M., Romero, I. A., Weksler, B., Couraud, P., & Drenckhahn, D. (2008). Differential effects of hydrocortisone and TNFα on tight junction proteins in an in vitro model of the human blood-brain barrier. The Journal of Physiology, 586(7), 1937-1949. doi:10.1113/jphysiol.2007.146852
- Nagasawa, K., Chiba, H., Fujita, H., Kojima, T., Saito, T., Endo, T., & Sawada, N. (2006). Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell. Physiol. Journal of Cellular Physiology, 208(1), 123-132. doi:10.1002/jcp.20647
- Marambaud, P., Dreses-Werringloer, U., & Vingtdeux, V. (2009). Calcium signaling in neurodegeneration. Molecular Neurodegeneration Mol Neurodegeneration, 4(1), 20. doi:10.1186/1750-1326-4-20
- Barnett MW, Larkman PM; Larkman (June 2007). "The action potential". Pract Neurol 7 (3): 192–7. PMID 17515599