Mechanism of autism
Autism's symptoms result from maturation-related changes in various systems of the brain. How autism occurs is not well understood. Its mechanism can be divided into two areas: the pathophysiology of brain structures and processes associated with autism, and the neuropsychological linkages between brain structures and behaviors.[1] The behaviors appear to have multiple pathophysiologies.[2][3]
There is evidence that gut–brain axis abnormalities may be involved.[4][5][6] A 2015 review proposed that immune dysregulation, gastrointestinal inflammation, malfunction of the autonomic nervous system, gut flora alterations, and food metabolites may cause brain neuroinflammation and dysfunction.[4] A 2016 review concludes that enteric nervous system abnormalities might play a role in neurological disorders such as autism. Neural connections and the immune system are a pathway that may allow diseases originated in the intestine to spread to the brain.[5]
Several lines of evidence point to synaptic dysfunction as a cause of autism.[7] Some rare mutations may lead to autism by disrupting some synaptic pathways, such as those involved with cell adhesion.[8] Gene replacement studies in mice suggest that autistic symptoms are closely related to later developmental steps that depend on activity in synapses and on activity-dependent changes.[9] All known teratogens (agents that cause birth defects) related to the risk of autism appear to act during the first eight weeks from conception, and though this does not exclude the possibility that autism can be initiated or affected later, there is strong evidence that autism arises very early in development.[10]
Pathophysiology
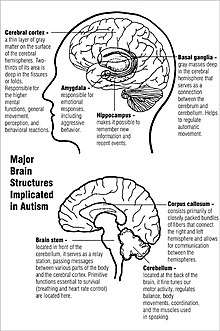
Unlike many other brain disorders, such as Parkinson's, autism does not have a clear unifying mechanism at either the molecular, cellular, or systems level; it is not known whether autism is a few disorders caused by mutations converging on a few common molecular pathways, or is (like intellectual disability) a large set of disorders with diverse mechanisms.[12] Autism appears to result from developmental factors that affect many or all functional brain systems,[13] and to disturb the timing of brain development more than the final product.[11] Neuroanatomical studies and the associations with teratogens strongly suggest that autism's mechanism includes alteration of brain development soon after conception.[10] This anomaly appears to start a cascade of pathological events in the brain that are significantly influenced by environmental factors.[14] Just after birth, the brains of children with autism tend to grow faster than usual, followed by normal or relatively slower growth in childhood. It is not known whether early overgrowth occurs in all children with autism. It seems to be most prominent in brain areas underlying the development of higher cognitive specialization.[15] Hypotheses for the cellular and molecular bases of pathological early overgrowth include the following:
- An excess of neurons that causes local overconnectivity in key brain regions.[16]
- Disturbed neuronal migration during early gestation.[17][18]
- Unbalanced excitatory–inhibitory networks.[18]
- Abnormal formation of synapses and dendritic spines,[18] for example, by modulation of the neurexin–neuroligin cell-adhesion system,[19] or by poorly regulated synthesis of synaptic proteins.[20][21] Disrupted synaptic development may also contribute to epilepsy, which may explain why the two conditions are associated.[22]
The immune system is thought to play an important role in autism. Children with autism have been found by researchers to have inflammation of both the peripheral and central immune systems as indicated by increased levels of pro-inflammatory cytokines and significant activation of microglia.[23][24][25] Biomarkers of abnormal immune function have also been associated with increased impairments in behaviors that are characteristic of the core features of autism such as, deficits in social interactions and communication.[24] Interactions between the immune system and the nervous system begin early during the embryonic stage of life, and successful neurodevelopment depends on a balanced immune response. It is thought that activation of a pregnant mother's immune system such as from environmental toxicants or infection can contribute to causing autism through causing a disruption of brain development.[26][27][28] This is supported by recent studies that have found that infection during pregnancy is associated with an increased risk of autism.[29][30]
The relationship of neurochemicals to autism is not well understood; several have been investigated, with the most evidence for the role of serotonin and of genetic differences in its transport.[7] The role of group I metabotropic glutamate receptors (mGluR) in the pathogenesis of fragile X syndrome, the most common identified genetic cause of autism, has led to interest in the possible implications for future autism research into this pathway.[31] Some data suggests neuronal overgrowth potentially related to an increase in several growth hormones[32] or to impaired regulation of growth factor receptors. Also, some inborn errors of metabolism are associated with autism, but probably account for less than 5% of cases.[33]
The mirror neuron system (MNS) theory of autism hypothesizes that distortion in the development of the MNS interferes with imitation and leads to autism's core features of social impairment and communication difficulties. The MNS operates when an animal performs an action or observes another animal perform the same action. The MNS may contribute to an individual's understanding of other people by enabling the modeling of their behavior via embodied simulation of their actions, intentions, and emotions.[34] Several studies have tested this hypothesis by demonstrating structural abnormalities in MNS regions of individuals with ASD, delay in the activation in the core circuit for imitation in individuals with Asperger syndrome, and a correlation between reduced MNS activity and severity of the syndrome in children with ASD.[35] However, individuals with autism also have abnormal brain activation in many circuits outside the MNS[36] and the MNS theory does not explain the normal performance of children with autism on imitation tasks that involve a goal or object.[37]
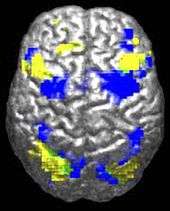
ASD-related patterns of low function and aberrant activation in the brain differ depending on whether the brain is doing social or nonsocial tasks.[39] In autism there is evidence for reduced functional connectivity of the default network (a large-scale brain network involved in social and emotional processing), with intact connectivity of the task-positive network (used in sustained attention and goal-directed thinking). In people with autism the two networks are not negatively correlated in time, suggesting an imbalance in toggling between the two networks, possibly reflecting a disturbance of self-referential thought.[40]
The underconnectivity theory of autism hypothesizes that autism is marked by underfunctioning high-level neural connections and synchronization, along with an excess of low-level processes.[41] Evidence for this theory has been found in functional neuroimaging studies on autistic individuals[42] and by a brainwave study that suggested that adults with ASD have local overconnectivity in the cortex and weak functional connections between the frontal lobe and the rest of the cortex.[43] Other evidence suggests the underconnectivity is mainly within each hemisphere of the cortex and that autism is a disorder of the association cortex.[44]
From studies based on event-related potentials, transient changes to the brain's electrical activity in response to stimuli, there is considerable evidence for differences in autistic individuals with respect to attention, orientation to auditory and visual stimuli, novelty detection, language and face processing, and information storage; several studies have found a preference for nonsocial stimuli.[45] For example, magnetoencephalography studies have found evidence in children with autism of delayed responses in the brain's processing of auditory signals.[46]
In the genetic area, relations have been found between autism and schizophrenia based on duplications and deletions of chromosomes; research showed that schizophrenia and autism are significantly more common in combination with 1q21.1 deletion syndrome. Research on autism/schizophrenia relations for chromosome 15 (15q13.3), chromosome 16 (16p13.1) and chromosome 17 (17p12) are inconclusive.[47]
Functional connectivity studies have found both hypo- and hyper-connectivity in brains of people with autism. Hypo-connectivity seems to dominate, especially for interhemispheric and cortico-cortical functional connectivity.[48]
Neuropsychology
Two major categories of cognitive theories have been proposed about the links between autistic brains and behavior.
The first category focuses on deficits in social cognition. Simon Baron-Cohen's empathizing–systemizing theory postulates that autistic individuals can systemize—that is, they can develop internal rules of operation to handle events inside the brain—but are less effective at empathizing by handling events generated by other agents. An extension, the extreme male brain theory, hypothesizes that autism is an extreme case of the male brain, defined psychometrically as individuals in whom systemizing is better than empathizing.[49] These theories are somewhat related to Baron-Cohen's earlier theory of mind approach, which hypothesizes that autistic behavior arises from an inability to ascribe mental states to oneself and others. The theory of mind hypothesis is supported by the atypical responses of children with autism to the Sally–Anne test for reasoning about others' motivations,[49] and the mirror neuron system theory of autism described in Pathophysiology maps well to the hypothesis.[35] However, most studies have found no evidence of impairment in autistic individuals' ability to understand other people's basic intentions or goals; instead, data suggests that impairments are found in understanding more complex social emotions or in considering others' viewpoints.[50]
The second category focuses on nonsocial or general processing: the executive functions such as working memory, planning, inhibition. In his review, Kenworthy states that "the claim of executive dysfunction as a causal factor in autism is controversial", however, "it is clear that executive dysfunction plays a role in the social and cognitive deficits observed in individuals with autism".[51] Tests of core executive processes such as eye movement tasks indicate improvement from late childhood to adolescence, but performance never reaches typical adult levels.[52] A strength of the theory is predicting stereotyped behavior and narrow interests;[53] two weaknesses are that executive function is hard to measure[51] and that executive function deficits have not been found in young children with autism.[54]
Weak central coherence theory hypothesizes that a limited ability to see the big picture underlies the central disturbance in autism. One strength of this theory is predicting special talents and peaks in performance in autistic people.[55] A related theory—enhanced perceptual functioning—focuses more on the superiority of locally oriented and perceptual operations in autistic individuals.[56] Yet another, monotropism, posits that autism stems from a different cognitive style, tending to focus attention (or processing resources) intensely, to the exclusion of other stimuli.[57] These theories map well from the underconnectivity theory of autism.
Neither category is satisfactory on its own; social cognition theories poorly address autism's rigid and repetitive behaviors, while most of the nonsocial theories have difficulty explaining social impairment and communication difficulties.[58] A combined theory based on multiple deficits may prove to be more useful.[59]
References
- Penn HE (2006). "Neurobiological correlates of autism: a review of recent research". Child Neuropsychol. 12 (1): 57–79. doi:10.1080/09297040500253546. PMID 16484102.
- London E (2007). "The role of the neurobiologist in redefining the diagnosis of autism". Brain Pathol. 17 (4): 408–11. doi:10.1111/j.1750-3639.2007.00103.x. PMID 17919126.
- Baird G, Cass H, Slonims V (2003). "Diagnosis of autism". BMJ. 327 (7413): 488–93. doi:10.1136/bmj.327.7413.488. PMC 188387. PMID 12946972.
- Wasilewska J, Klukowski M (2015). "Gastrointestinal symptoms and autism spectrum disorder: links and risks - a possible new overlap syndrome". Pediatric Health Med Ther (Review). 6: 153–166. doi:10.2147/PHMT.S85717. PMC 5683266. PMID 29388597.
- Rao M, Gershon MD (September 2016). "The bowel and beyond: the enteric nervous system in neurological disorders". Nat Rev Gastroenterol Hepatol (Review). 13 (9): 517–28. doi:10.1038/nrgastro.2016.107. PMC 5005185. PMID 27435372.
- Israelyan N, Margolis KG (2018). "Serotonin as a link between the gut-brain-microbiome axis in autism spectrum disorders". Pharmacol Res (Review). 132: 1–6. doi:10.1016/j.phrs.2018.03.020. PMC 6368356. PMID 29614380.
- Levy SE, Mandell DS, Schultz RT (2009). "Autism". Lancet. 374 (9701): 1627–38. doi:10.1016/S0140-6736(09)61376-3. PMC 2863325. PMID 19819542.
- Betancur C, Sakurai T, Buxbaum JD (2009). "The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders". Trends Neurosci. 32 (7): 402–12. doi:10.1016/j.tins.2009.04.003. PMID 19541375.
- Walsh CA, Morrow EM, Rubenstein JL (2008). "Autism and brain development". Cell. 135 (3): 396–400. doi:10.1016/j.cell.2008.10.015. PMC 2701104. PMID 18984148.
- Arndt TL, Stodgell CJ, Rodier PM (2005). "The teratology of autism". Int J Dev Neurosci. 23 (2–3): 189–99. doi:10.1016/j.ijdevneu.2004.11.001. PMID 15749245.
- Amaral DG, Schumann CM, Nordahl CW (2008). "Neuroanatomy of autism". Trends Neurosci. 31 (3): 137–45. doi:10.1016/j.tins.2007.12.005. PMID 18258309.
- Geschwind DH (2008). "Autism: many genes, common pathways?". Cell. 135 (3): 391–95. doi:10.1016/j.cell.2008.10.016. PMC 2756410. PMID 18984147.
- Müller RA (2007). "The study of autism as a distributed disorder". Ment Retard Dev Disabil Res Rev. 13 (1): 85–95. doi:10.1002/mrdd.20141. PMC 3315379. PMID 17326118.
- Casanova MF (2007). "The neuropathology of autism". Brain Pathol. 17 (4): 422–33. doi:10.1111/j.1750-3639.2007.00100.x. PMID 17919128.
- Geschwind DH (2009). "Advances in autism". Annu Rev Med. 60: 367–80. doi:10.1146/annurev.med.60.053107.121225. PMC 3645857. PMID 19630577.
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, Morgan J (2007). "Mapping early brain development in autism". Neuron. 56 (2): 399–413. doi:10.1016/j.neuron.2007.10.016. PMID 17964254.
- Schmitz C, Rezaie P (2008). "The neuropathology of autism: where do we stand?". Neuropathol Appl Neurobiol. 34 (1): 4–11. doi:10.1111/j.1365-2990.2007.00872.x. PMID 17971078.
- Persico AM, Bourgeron T (2006). "Searching for ways out of the autism maze: genetic, epigenetic and environmental clues". Trends Neurosci. 29 (7): 349–58. doi:10.1016/j.tins.2006.05.010. PMID 16808981.
- Südhof TC (2008). "Neuroligins and neurexins link synaptic function to cognitive disease". Nature. 455 (7215): 903–11. Bibcode:2008Natur.455..903S. doi:10.1038/nature07456. PMC 2673233. PMID 18923512.
- Kelleher RJ, Bear MF (2008). "The autistic neuron: troubled translation?". Cell. 135 (3): 401–06. doi:10.1016/j.cell.2008.10.017. PMID 18984149.
- Bear MF, Dölen G, Osterweil E, Nagarajan N (2008). "Fragile X: translation in action". Neuropsychopharmacology. 33 (1): 84–7. doi:10.1038/sj.npp.1301610. PMC 4327813. PMID 17940551.
- Tuchman R, Moshé SL, Rapin I (2009). "Convulsing toward the pathophysiology of autism". Brain Dev. 31 (2): 95–103. doi:10.1016/j.braindev.2008.09.009. PMC 2734903. PMID 19006654.
- Hsiao EY (2013). "Immune Dysregulation in Autism Spectrum Disorder". Neurobiology of Autism. International Review of Neurobiology. 113. pp. 269–302. doi:10.1016/B978-0-12-418700-9.00009-5. ISBN 9780124187009. PMID 24290389.
- Onore C, Careaga M, Ashwood P (August 2011). "The role of immune dysfunction in the pathophysiology of autism". Brain, Behavior, and Immunity. 26 (3): 383–92. doi:10.1016/j.bbi.2011.08.007. PMC 3418145. PMID 21906670.
- Rossignol DA, Frye RE (2014). "Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism". Frontiers in Physiology. 5: 150. doi:10.3389/fphys.2014.00150. PMC 4001006. PMID 24795645.
- Patterson PH (July 2011). "Maternal infection and immune involvement in autism". Trends in Molecular Medicine. 17 (7): 389–94. doi:10.1016/j.molmed.2011.03.001. PMC 3135697. PMID 21482187.
- Chaste P, Leboyer M (2012). "Autism risk factors: genes, environment, and gene-environment interactions". Dialogues Clin Neurosci. 14 (3): 281–92. PMC 3513682. PMID 23226953.
- Ashwood P, Wills S, Van de Water J (2006). "The immune response in autism: a new frontier for autism research". J Leukoc Biol. 80 (1): 1–15. CiteSeerX 10.1.1.329.777. doi:10.1189/jlb.1205707. PMID 16698940. Archived from the original on 5 October 2006.
- Lee BK, Magnusson C, Gardner RM, Blomström S, Newschaffer CJ, Burstyn I, Karlsson H, Dalman C (September 2014). "Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders". Brain, Behavior, and Immunity. 44: 100–105. doi:10.1016/j.bbi.2014.09.001. PMC 4418173. PMID 25218900.
- Atladóttir HO, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET (December 2010). "Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders". Journal of Autism and Developmental Disorders. 40 (12): 1423–30. doi:10.1007/s10803-010-1006-y. PMID 20414802.
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF (2007). "Correction of fragile X syndrome in mice". Neuron. 56 (6): 955–62. doi:10.1016/j.neuron.2007.12.001. PMC 2199268. PMID 18093519.
- Hughes JR (2009). "Update on autism: A review of 1300 reports published in 2008". Epilepsy Behav. 16 (4): 569–89. doi:10.1016/j.yebeh.2009.09.023. PMID 19896907.
- Manzi B, Loizzo AL, Giana G, Curatolo P (2008). "Autism and metabolic diseases". J Child Neurol. 23 (3): 307–14. doi:10.1177/0883073807308698. PMID 18079313.
- MNS and autism:
- Williams JH (2008). "Self–other relations in social development and autism: multiple roles for mirror neurons and other brain bases". Autism Res. 1 (2): 73–90. doi:10.1002/aur.15. PMID 19360654.
- Dinstein I, Thomas C, Behrmann M, Heeger DJ (2008). "A mirror up to nature". Curr Biol. 18 (1): R13–18. doi:10.1016/j.cub.2007.11.004. PMC 2517574. PMID 18177704.
- Iacoboni M, Dapretto M (2006). "The mirror neuron system and the consequences of its dysfunction". Nature Reviews Neuroscience. 7 (12): 942–51. doi:10.1038/nrn2024. PMID 17115076.
- Frith U, Frith CD (2003). "Development and neurophysiology of mentalizing". Philosophical Transactions of the Royal Society B. 358 (1431): 459–73. doi:10.1098/rstb.2002.1218. PMC 1693139. PMID 12689373.
- Hamilton AF (2008). "Emulation and mimicry for social interaction: a theoretical approach to imitation in autism". Q J Exp Psychol. 61 (1): 101–15. doi:10.1080/17470210701508798. PMID 18038342.
- Powell K (2004). "Opening a window to the autistic brain". PLoS Biol. 2 (8): E267. doi:10.1371/journal.pbio.0020267. PMC 509312. PMID 15314667.
- Di Martino A, Ross K, Uddin LQ, Sklar AB, Castellanos FX, Milham MP (2009). "Functional brain correlates of social and nonsocial processes in autism spectrum disorders: an activation likelihood estimation meta-analysis". Biol Psychiatry. 65 (1): 63–74. doi:10.1016/j.biopsych.2008.09.022. PMC 2993772. PMID 18996505.
- Broyd SJ, Demanuele C, Debener S, Helps SK, James CJ, Sonuga-Barke EJ (2009). "Default-mode brain dysfunction in mental disorders: a systematic review". Neurosci Biobehav Rev. 33 (3): 279–96. doi:10.1016/j.neubiorev.2008.09.002. PMID 18824195.
- Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ (2007). "Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry". Cereb Cortex. 17 (4): 951–61. doi:10.1093/cercor/bhl006. PMC 4500121. PMID 16772313. Archived from the original on 7 July 2010.
- Williams DL, Goldstein G, Minshew NJ (2006). "Neuropsychologic functioning in children with autism: further evidence for disordered complex information-processing". Child Neuropsychol. 12 (4–5): 279–98. doi:10.1080/09297040600681190. PMC 1803025. PMID 16911973.
- Murias M, Webb SJ, Greenson J, Dawson G (2007). "Resting state cortical connectivity reflected in EEG coherence in individuals with autism". Biol Psychiatry. 62 (3): 270–73. doi:10.1016/j.biopsych.2006.11.012. PMC 2001237. PMID 17336944.
- Minshew NJ, Williams DL (2007). "The new neurobiology of autism: cortex, connectivity, and neuronal organization". Arch Neurol. 64 (7): 945–50. doi:10.1001/archneur.64.7.945. PMC 2597785. PMID 17620483.
- Jeste SS, Nelson CA (2009). "Event related potentials in the understanding of autism spectrum disorders: an analytical review". J Autism Dev Disord. 39 (3): 495–510. doi:10.1007/s10803-008-0652-9. PMC 4422389. PMID 18850262.
- Roberts TP, Schmidt GL, Egeth M, Blaskey L, Rey MM, Edgar JC, Levy SE (2008). "Electrophysiological signatures: magnetoencephalographic studies of the neural correlates of language impairment in autism spectrum disorders". Int J Psychophysiol. 68 (2): 149–60. doi:10.1016/j.ijpsycho.2008.01.012. PMC 2397446. PMID 18336941.
- Crespi B, Stead P, Elliot M (2010). "Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia". Proceedings of the National Academy of Sciences of the United States of America. 107 (Suppl 1): 1736–41. Bibcode:2010PNAS..107.1736C. doi:10.1073/pnas.0906080106. PMC 2868282. PMID 19955444.
- Ha S, Sohn IJ, Kim N, Sim HJ, Cheon KA (December 2015). "Characteristics of Brains in Autism Spectrum Disorder: Structure, Function and Connectivity across the Lifespan". Exp Neurobiol (Review). 24 (4): 273–84. doi:10.5607/en.2015.24.4.273. PMC 4688328. PMID 26713076.
- Baron-Cohen S (2009). "Autism: the empathizing–systemizing (E-S) theory" (PDF). Annals of the New York Academy of Sciences. 1156 (1): 68–80. doi:10.1111/j.1749-6632.2009.04467.x. PMID 19338503.
- Hamilton AF (2009). "Goals, intentions and mental states: challenges for theories of autism". J Child Psychol Psychiatry. 50 (8): 881–92. CiteSeerX 10.1.1.621.6275. doi:10.1111/j.1469-7610.2009.02098.x. PMID 19508497.
- Kenworthy L, Yerys BE, Anthony LG, Wallace GL (2008). "Understanding executive control in autism spectrum disorders in the lab and in the real world". Neuropsychol Rev. 18 (4): 320–38. doi:10.1007/s11065-008-9077-7. PMC 2856078. PMID 18956239.
- O'Hearn K, Asato M, Ordaz S, Luna B (2008). "Neurodevelopment and executive function in autism". Dev Psychopathol. 20 (4): 1103–32. doi:10.1017/S0954579408000527. PMID 18838033.
- Hill EL (2004). "Executive dysfunction in autism". Trends Cogn Sci. 8 (1): 26–32. doi:10.1016/j.dr.2004.01.001. PMID 14697400.
- Sigman M, Spence SJ, Wang AT (2006). "Autism from developmental and neuropsychological perspectives". Annual Review of Clinical Psychology. 2: 327–55. doi:10.1146/annurev.clinpsy.2.022305.095210. PMID 17716073.
- Happé F, Frith U (January 2006). "The weak coherence account: detail-focused cognitive style in autism spectrum disorders". Journal of Autism and Developmental Disorders. 36 (1): 5–25. doi:10.1007/s10803-005-0039-0. PMID 16450045.
- Mottron L, Dawson M, Soulières I, Hubert B, Burack J (January 2006). "Enhanced perceptual functioning in autism: an update, and eight principles of autistic perception". Journal of Autism and Developmental Disorders. 36 (1): 27–43. doi:10.1007/s10803-005-0040-7. PMID 16453071.
- Murray D, Lesser M, Lawson W (May 2005). "Attention, monotropism and the diagnostic criteria for autism" (PDF). Autism. 9 (2): 139–56. doi:10.1177/1362361305051398. PMID 15857859. Archived from the original (PDF) on 19 May 2018. Retrieved 18 March 2018.
- Happé F, Ronald A, Plomin R (2006). "Time to give up on a single explanation for autism". Nature Neuroscience. 9 (10): 1218–20. doi:10.1038/nn1770. PMID 17001340.
- Rajendran G, Mitchell P (2007). "Cognitive theories of autism" (PDF). Dev Rev. 27 (2): 224–60. doi:10.1016/j.dr.2007.02.001.