Gaseous signaling molecules
Gaseous signaling molecules are gaseous molecules that are either synthesised internally (endogenously) in the organism, tissue or cell or are received by the organism, tissue or cell from outside (say, from the atmosphere or hydrosphere, as in the case of oxygen) and that are used to transmit chemical signals which induce certain physiological or biochemical changes in the organism, tissue or cell. The term is applied to, for example, oxygen, carbon dioxide, nitric oxide, carbon monoxide, hydrogen sulfide, sulfur dioxide, nitrous oxide, hydrogen cyanide, ammonia, methane, hydrogen, ethylene, etc.
Many, but not all, gaseous signaling molecules are called gasotransmitters.
The biological roles of each of the gaseous signaling molecules are in short outlined below.
Gaseous signaling molecules as gasotransmitters
Gasotransmitters is a subfamily of endogenous molecules of gases or gaseous signaling molecules, including NO, CO, H
2S.[1] These particular gases share many common features in their production and function but carry on their tasks in unique ways, which differ from classical signaling molecules, in the human body. In 1981, it was first suggested from clinical work with nitrous oxide that a gas had a direct action at pharmacological receptors and thereby acted as a neurotransmitter.[2][3][4] In vitro experiments confirmed these observations[5] which were replicated at NIDA later.[6]
The terminology and characterization criteria of “gasotransmitter” were firstly introduced in 2002.[7] For one gas molecule to be categorized as a gasotransmitters, all of the following criteria should be met.[8][7]
- It is a small molecule of gas;
- It is freely permeable to membranes. As such, its effects do not rely on the cognate membrane receptors. It can have endocrine, paracrine, and autocrine effects. In their endocrine mode of action, for example, gasotransmitters can enter the blood stream; be carried to remote targets by scavengers and released there, and modulate functions of remote target cells;
- It is endogenously and enzymatically generated and its production is regulated;
- It has well defined and specific functions at physiologically relevant concentrations. Thus, manipulating the endogenous levels of this gas evokes specific physiological changes;
- Functions of this endogenous gas can be mimicked by its exogenously applied counterpart;
- Its cellular effects may or may not be mediated by second messengers, but should have specific cellular and molecular targets.
In 2011, a European Network on Gasotransmitters (ENOG) was formed. The aim of the network is to promote research on NO, CO and H
2S in order to better understand the biology of gasotransmitters and to unravel the role of each mediator in health and disease. Moreover, the network aims to contribute to the translation of basic science knowledge in this area of research into therapeutic or diagnostic tools.
Oxygen
Carbon dioxide
Carbon dioxide is one of the mediators of local autoregulation of blood supply. If its levels are high, the capillaries expand to allow a greater blood flow to that tissue.
Bicarbonate ions are crucial for regulating blood pH. A person's breathing rate influences the level of CO2 in their blood. Breathing that is too slow or shallow causes respiratory acidosis, while breathing that is too rapid leads to hyperventilation, which can cause respiratory alkalosis.
Although the body requires oxygen for metabolism, low oxygen levels normally do not stimulate breathing. Rather, breathing is stimulated by higher carbon dioxide levels.
The respiratory centers try to maintain an arterial CO2 pressure of 40 mm Hg. With intentional hyperventilation, the CO2 content of arterial blood may be lowered to 10–20 mm Hg (the oxygen content of the blood is little affected), and the respiratory drive is diminished. This is why one can hold one's breath longer after hyperventilating than without hyperventilating. This carries the risk that unconsciousness may result before the need to breathe becomes overwhelming, which is why hyperventilation is particularly dangerous before free diving.
Nitric oxide
NO is one of the few gaseous signalling molecules known and is additionally exceptional due to the fact that it is a radical gas. It is a key vertebrate biological messenger, playing a role in a variety of biological processes.[10] It is a known bioproduct in almost all types of organisms, ranging from bacteria to plants, fungi, and animal cells.[11]
Nitric oxide, known as the 'endothelium-derived relaxing factor', or 'EDRF', is biosynthesized endogenously from L-arginine, oxygen, and NADPH by various nitric oxide synthase (NOS) enzymes. Reduction of inorganic nitrate may also serve to make nitric oxide. The endothelium (inner lining) of blood vessels uses nitric oxide to signal the surrounding smooth muscle to relax, thus resulting in vasodilation and increasing blood flow. Nitric oxide is highly reactive (having a lifetime of a few seconds), yet diffuses freely across membranes. These attributes make nitric oxide ideal for a transient paracrine (between adjacent cells) and autocrine (within a single cell) signaling molecule.[12]
Independent of nitric oxide synthase, an alternative pathway, coined the nitrate-nitrite-nitric oxide pathway, elevates nitric oxide through the sequential reduction of dietary nitrate derived from plant-based foods.[13] Nitrate-rich vegetables, in particular leafy greens, such as spinach and arugula, and beetroot, have been shown to increase cardioprotective levels of nitric oxide with a corresponding reduction in blood pressure in pre-hypertensive persons.[14][15] For the body to generate nitric oxide through the nitrate-nitrite-nitric oxide pathway, the reduction of nitrate to nitrite occurs in the mouth, by commensal bacteria, an obligatory and necessary step.[16] Monitoring nitric oxide status by saliva testing detects the bioconversion of plant-derived nitrate into nitric oxide. A rise in salivary levels is indicative of diets rich in leafy vegetables which are often abundant in anti-hypertensive diets such as the DASH diet.[17]
The production of nitric oxide is elevated in populations living at high altitudes, which helps these people avoid hypoxia by aiding in pulmonary vasculature vasodilation. Effects include vasodilatation, neurotransmission, modulation of the hair cycle,[18] production of reactive nitrogen intermediates and penile erections (through its ability to vasodilate). Nitroglycerin and amyl nitrite serve as vasodilators because they are converted to nitric oxide in the body. The vasodilating antihypertensive drug minoxidil contains an NO moiety and may act as an NO agonist. Likewise, Sildenafil citrate, popularly known by the trade name Viagra, stimulates erections primarily by enhancing signaling through the nitric oxide pathway in the penis.
Nitric oxide (NO) contributes to vessel homeostasis by inhibiting vascular smooth muscle contraction and growth, platelet aggregation, and leukocyte adhesion to the endothelium. Humans with atherosclerosis, diabetes, or hypertension often show impaired NO pathways.[19] A high salt intake was demonstrated to attenuate NO production in patients with essential hypertension, although bioavailability remains unregulated.[20]
Nitric oxide is also generated by phagocytes (monocytes, macrophages, and neutrophils) as part of the human immune response.[21] Phagocytes are armed with inducible nitric oxide synthase (iNOS), which is activated by interferon-gamma (IFN-γ) as a single signal or by tumor necrosis factor (TNF) along with a second signal.[22][23][24] On the other hand, transforming growth factor-beta (TGF-β) provides a strong inhibitory signal to iNOS, whereas interleukin-4 (IL-4) and IL-10 provide weak inhibitory signals. In this way, the immune system may regulate the resources of phagocytes that play a role in inflammation and immune responses.[25] Nitric oxide is secreted as free radicals in an immune response and is toxic to bacteria and intracellular parasites, including Leishmania[26] and malaria;[27][28][29] the mechanism for this includes DNA damage[30][31][32] and degradation of iron sulfur centers into iron ions and iron-nitrosyl compounds.[33]
In response, many bacterial pathogens have evolved mechanisms for nitric oxide resistance.[34] Because nitric oxide might have proinflammatory actions in conditions like asthma, there has been increasing interest in the use of exhaled nitric oxide as a breath test in diseases with airway inflammation. Reduced levels of exhaled NO have been associated with exposure to air pollution in cyclists and smokers, but, in general, increased levels of exhaled NO are associated with exposure to air pollution.[35]
Nitric oxide can contribute to reperfusion injury when an excessive amount produced during reperfusion (following a period of ischemia) reacts with superoxide to produce the damaging oxidant peroxynitrite. In contrast, inhaled nitric oxide has been shown to help survival and recovery from paraquat poisoning, which produces lung tissue-damaging superoxide and hinders NOS metabolism.
In plants, nitric oxide can be produced by any of four routes: (i) L-arginine-dependent nitric oxide synthase,[36][37][38] (although the existence of animal NOS homologs in plants is debated),[39] (ii) plasma membrane-bound nitrate reductase, (iii) mitochondrial electron transport chain, or (iv) non-enzymatic reactions. It is a signaling molecule, acts mainly against oxidative stress and also plays a role in plant pathogen interactions. Treating cut flowers and other plants with nitric oxide has been shown to lengthen the time before wilting.[40]
Two important biological reaction mechanisms of nitric oxide are S-nitrosation of thiols, and nitrosylation of transition metal ions. S-nitrosation involves the (reversible) conversion of thiol groups, including cysteine residues in proteins, to form S-nitrosothiols (RSNOs). S-Nitrosation is a mechanism for dynamic, post-translational regulation of most or all major classes of protein.[41] The second mechanism, nitrosylation, involves the binding of NO to a transition metal ion like iron or copper. In this function, NO is referred to as a nitrosyl ligand. Typical cases involve the nitrosylation of heme proteins like cytochromes, thereby disabling the normal enzymatic activity of the enzyme. Nitrosylated ferrous iron is particularly stable, as the binding of the nitrosyl ligand to ferrous iron (Fe(II)) is very strong. Hemoglobin is a prominent example of a heme protein that may be modified by NO by both pathways: NO may attach directly to the heme in the nitrosylation reaction, and independently form S-nitrosothiols by S-nitrosation of the thiol moieties.[42]
There are several mechanisms by which NO has been demonstrated to affect the biology of living cells. These include oxidation of iron-containing proteins such as ribonucleotide reductase and aconitase, activation of the soluble guanylate cyclase, ADP ribosylation of proteins, protein sulfhydryl group nitrosylation, and iron regulatory factor activation.[43] NO has been demonstrated to activate NF-κB in peripheral blood mononuclear cells, an important transcription factor in iNOS gene expression in response to inflammation.[44]
It was found that NO acts through the stimulation of the soluble guanylate cyclase, which is a heterodimeric enzyme with subsequent formation of cyclic-GMP. Cyclic-GMP activates protein kinase G, which causes reuptake of Ca2+ and the opening of calcium-activated potassium channels. The fall in concentration of Ca2+ ensures that the myosin light-chain kinase (MLCK) can no longer phosphorylate the myosin molecule, thereby stopping the crossbridge cycle and leading to relaxation of the smooth muscle cell.[45]
Nitrous oxide
Nitrous oxide in biological systems can be formed by an enzymatic or non-enzymatic reduction of nitric oxide.[46] In vitro studies have shown that endogenous nitrous oxide can be formed by the reaction between nitric oxide and thiol.[47] Some authors have shown that this process of NO reduction to N2O takes place in hepatocytes, specifically in their cytoplasm and mitochondria, and suggested that the N2O can possibly be produced in mammalian cells.[48] It is well known that N2O is produced by some bacteria during process called denitrification.
Apart from its direct[5][6] and indirect actions at opioid receptors,[49] it was also shown that N2O inhibits NMDA receptor-mediated activity and ionic currents and diminishes NMDA receptor-mediated excitotoxicity and neurodegeneration.[50] Nitrous oxide also inhibits methionine synthase and slows the conversion of homocysteine to methionine, increases homocysteine concentration and decreases methionine concentration. This effect was shown in lymphocyte cell cultures[51] and in human liver biopsy samples.[52]
Nitrous oxide does not bind as a ligand to the heme and does not react with thiol-containing proteins. Nevertheless, studies have shown that nitrous oxide can reversibly and non-covalently "insert" itself into the inner structures of some heme-containing proteins such as hemoglobin, myoglobin, cytochrome oxidase and alter their structure and function.[53] The ability of nitrous oxide to alter the structure and function of these proteins was demonstrated by shifts in infrared spectra of cysteine thiols of hemoglobin[54] and by partial and reversible inhibition of cytochrome oxidase.[55]
Endogenous nitrous oxide can possibly play a role in modulating endogenous opioid[56][2] and NMDA systems.[50]
Carbon monoxide
Carbon monoxide is produced naturally by the human body as a signaling molecule. Thus, carbon monoxide may have a physiological role in the body, such as a neurotransmitter or a blood vessel relaxant.[57] Because of carbon monoxide's role in the body, abnormalities in its metabolism have been linked to a variety of diseases, including neurodegenerations, hypertension, heart failure, and inflammation.[57]
Summary of function:[58]
- CO functions as an endogenous signaling molecule.
- CO modulates functions of the cardiovascular system.
- CO inhibits blood platelet aggregation and adhesion.
- CO may play a role as potential therapeutic agent.
In mammals, carbon monoxide is naturally produced by the action of heme oxygenase 1 and 2 on the heme from hemoglobin breakdown. This process produces a certain amount of carboxyhemoglobin in normal persons, even if they do not breathe any carbon monoxide.
Following the first report that carbon monoxide is a normal neurotransmitter in 1993,[59][60] as well as one of three gases that naturally modulate inflammatory responses in the body (the other two being nitric oxide and hydrogen sulfide), carbon monoxide has received a great deal of clinical attention as a biological regulator. In many tissues, all three gases are known to act as anti-inflammatories, vasodilators, and encouragers of neovascular growth.[61] However, the issues are complex, as neovascular growth is not always beneficial, since it plays a role in tumor growth, and also the damage from wet macular degeneration, a disease for which smoking (a major source of carbon monoxide in the blood, several times more than natural production) increases the risk from 4 to 6 times.
There is a theory that, in some nerve cell synapses, when long-term memories are being laid down, the receiving cell makes carbon monoxide, which back-transmits to the transmitting cell, telling it to transmit more readily in future. Some such nerve cells have been shown to contain guanylate cyclase, an enzyme that is activated by carbon monoxide.[60]
Studies involving carbon monoxide have been conducted in many laboratories throughout the world for its anti-inflammatory and cytoprotective properties. These properties have potential to be used to prevent the development of a series of pathological conditions including ischemia reperfusion injury, transplant rejection, atherosclerosis, severe sepsis, severe malaria, or autoimmunity. Clinical tests involving humans have been performed, however the results have not yet been released.[62]
Carbon suboxide
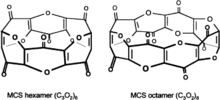
Carbon suboxide, C3O2, can be produced in small amounts in any biochemical process that normally produces carbon monoxide, CO, for example, during heme oxidation by heme oxygenase-1. It can also be formed from malonic acid. It has been shown that carbon suboxide in an organism can quickly polymerize into macrocyclic polycarbon structures with the common formula (C3O2)n (mostly (C3O2)6 and (C3O2)8), and that those macrocyclic compounds are potent inhibitors of Na+/K+-ATP-ase and Ca-dependent ATP-ase, and have digoxin-like physiological properties and natriuretic and antihypertensive actions. Those macrocyclic carbon suboxide polymer compounds are thought to be endogenous digoxin-like regulators of Na+/K+-ATP-ases and Ca-dependent ATP-ases, and endogenous natriuretics and antihypertensives.[63][64][65] Other than that, some authors think also that those macrocyclic compounds of carbon suboxide can possibly diminish free radical formation and oxidative stress and play a role in endogenous anticancer protective mechanisms, for example in the retina.[66]
Hydrogen sulfide
Hydrogen sulfide is produced in small amounts by some cells of the mammalian body and has a number of biological signaling functions. (Only two other such gases are currently known: nitric oxide (NO) and carbon monoxide (CO).)
The gas is produced from cysteine by the enzymes cystathionine beta-synthase and cystathionine gamma-lyase. It acts as a relaxant of smooth muscle and as a vasodilator[67] and is also active in the brain, where it increases the response of the NMDA receptor and facilitates long term potentiation,[68] which is involved in the formation of memory.
Eventually the gas is converted to sulfite in the mitochondria by thiosulfate reductase, and the sulfite is further oxidized to thiosulfate and sulfate by sulfite oxidase. The sulfates are excreted in the urine.[69]
Due to its effects similar to nitric oxide (without its potential to form peroxides by interacting with superoxide), hydrogen sulfide is now recognized as potentially protecting against cardiovascular disease.[67] The cardioprotective role effect of garlic is caused by catabolism of the polysulfide group in allicin to H
2S, a reaction that could depend on reduction mediated by glutathione.[70]
Though both nitric oxide (NO) and hydrogen sulfide have been shown to relax blood vessels, their mechanisms of action are different: while NO activates the enzyme guanylyl cyclase, H
2S activates ATP-sensitive potassium channels in smooth muscle cells. Researchers are not clear how the vessel-relaxing responsibilities are shared between nitric oxide and hydrogen sulfide. However, there exists some evidence to suggest that nitric oxide does most of the vessel-relaxing work in large vessels and hydrogen sulfide is responsible for similar action in smaller blood vessels.[71]
Recent findings suggest strong cellular crosstalk of NO and H
2S,[72] demonstrating that the vasodilatatory effects of these two gases are mutually dependent. Additionally, H
2S reacts with intracellular S-nitrosothiols to form the smallest S-nitrosothiol (HSNO), and a role of hydrogen sulfide in controlling the intracellular S-nitrosothiol pool has been suggested.[73]
Like nitric oxide, hydrogen sulfide is involved in the relaxation of smooth muscle that causes erection of the penis, presenting possible new therapy opportunities for erectile dysfunction.[74][75]
Hydrogen sulfide (H
2S) deficiency can be detrimental to the vascular function after an acute myocardial infarction (AMI).[76] AMIs can lead to cardiac dysfunction through two distinct changes; increased oxidative stress via free radical accumulation and decreased NO bioavailability.[77] Free radical accumulation occurs due to increased electron transport uncoupling at the active site of endothelial nitric oxide synthase (eNOS), an enzyme involved in converting L-arginine to NO.[76][77] During an AMI, oxidative degradation of tetrahydrobiopterin (BH4), a cofactor in NO production, limits BH4 availability and limits NO productionby eNOS.[77] Instead, eNOS reacts with oxygen, another cosubstrates involved in NO production. The products of eNOS are reduced to superoxides, increasing free radical production and oxidative stress within the cells.[76] A H
2S deficiency impairs eNOS activity by limiting Akt activation and inhibiting Akt phosphorylation of the eNOSS1177 activation site.[76][72] Instead, Akt activity is increased to phosphorylate the eNOST495 inhibition site, downregulating eNOS production of NO.[76][72]
H
2S therapy uses a H
2S donor, such as diallyl trisulfide (DATS), to increase the supply of H
2S to an AMI patient. H
2S donors reduce myocardial injury and reperfusion complications.[76] Increased H
2S levels within the body will react with oxygen to produce sulfane sulfur, a storage intermediate for H
2S.[76] H
2S pools in the body attracts oxygen to react with excess H
2S and eNOS to increase NO production.[76] With increased use of oxygen to produce more NO, less oxygen is available to react with eNOS to produce superoxides during an AMI, ultimately lowering the accumulation of reactive oxygen species (ROS).[76] Furthermore, decreased accumulation of ROS lowers oxidative stress in vascular smooth muscle cells, decreasing oxidative degeneration of BH4.[77] Increased BH4 cofactor contributes to increased production of NO within the body.[77] Higher concentrations of H
2S directly increase eNOS activity through Akt activation to increase phosphorylation of the eNOSS1177 activation site, and decrease phosphorylation of the eNOST495 inhibition site.[76][72] This phosphorylation process upregulates eNOS activity, catalyzing more conversion of L-arginine to NO.[76][72] Increased NO production enables soluble guanylyl cyclase (sGC) activity, leading to an increased conversion of guanosine triphosphate (GTP) to 3’,5’-cyclic guanosine monophosphate (cGMP).[78] In H
2S therapy immediately following an AMI, increased cGMP triggers an increase in protein kinase G (PKG) activity.[79] PKG reduces intracellular Ca2+ in vascular smooth muscle to increase smooth muscle relaxation and promote blood flow.[79] PKG also limits smooth muscle cell proliferation, reducing intima thickening following AMI injury, ultimately decreasing myocardial infarct size.[76][78]
In Alzheimer's disease the brain's hydrogen sulfide concentration is severely decreased.[80] In a certain rat model of Parkinson's disease, the brain's hydrogen sulfide concentration was found to be reduced, and administering hydrogen sulfide alleviated the condition.[81] In trisomy 21 (Down syndrome) the body produces an excess of hydrogen sulfide.[69] Hydrogen sulfide is also involved in the disease process of type 1 diabetes. The beta cells of the pancreas in type 1 diabetes produce an excess of the gas, leading to the death of these cells and to a reduced production of insulin by those that remain.[71]
In 2005, it was shown that mice can be put into a state of suspended animation-like hypothermia by applying a low dosage of hydrogen sulfide (81 ppm H
2S) in the air. The breathing rate of the animals sank from 120 to 10 breaths per minute and their temperature fell from 37 °C to just 2 °C above ambient temperature (in effect, they had become cold-blooded). The mice survived this procedure for 6 hours and afterwards showed no negative health consequences.[82] In 2006 it was shown that the blood pressure of mice treated in this fashion with hydrogen sulfide did not significantly decrease.[83]
A similar process known as hibernation occurs naturally in many mammals and also in toads, but not in mice. (Mice can fall into a state called clinical torpor when food shortage occurs). If the H
2S-induced hibernation can be made to work in humans, it could be useful in the emergency management of severely injured patients, and in the conservation of donated organs. In 2008, hypothermia induced by hydrogen sulfide for 48 hours was shown to reduce the extent of brain damage caused by experimental stroke in rats.[84]
As mentioned above, hydrogen sulfide binds to cytochrome oxidase and thereby prevents oxygen from binding, which leads to the dramatic slowdown of metabolism. Animals and humans naturally produce some hydrogen sulfide in their body; researchers have proposed that the gas is used to regulate metabolic activity and body temperature, which would explain the above findings.[85]
Two recent studies cast doubt that the effect can be achieved in larger mammals. A 2008 study failed to reproduce the effect in pigs, concluding that the effects seen in mice were not present in larger mammals.[86] Likewise a paper by Haouzi et al. noted that there is no induction of hypometabolism in sheep, either.[87]
At the February 2010 TED conference, Mark Roth announced that hydrogen sulfide induced hypothermia in humans had completed Phase I clinical trials.[88] The clinical trials commissioned by the company he helped found, Ikaria, were however withdrawn or terminated by August 2011.[89][90]
Sulfur dioxide
The role of sulfur dioxide in mammalian biology is not yet well understood.[91] Sulfur dioxide blocks nerve signals from the pulmonary stretch receptors and abolishes the Hering–Breuer inflation reflex.
It was shown that endogenous sulfur dioxide plays a role in diminishing an experimental lung damage caused by oleic acid. Endogenous sulfur dioxide lowered lipid peroxidation, free radical formation, oxidative stress and inflammation during an experimental lung damage. Conversely, a successful lung damage caused a significant lowering of endogenous sulfur dioxide production, and an increase in lipid peroxidation, free radical formation, oxidative stress and inflammation. Moreover, blockade of an enzyme that produces endogenous SO2 significantly increased the amount of lung tissue damage in the experiment. Conversely, adding acetylcysteine or glutathione to the rat diet increased the amount of endogenous SO2 produced and decreased the lung damage, the free radical formation, oxidative stress, inflammation and apoptosis.[92]
It is considered that endogenous sulfur dioxide plays a significant physiological role in regulating cardiac and blood vessel function, and aberrant or deficient sulfur dioxide metabolism can contribute to several different cardiovascular diseases, such as arterial hypertension, atherosclerosis, pulmonary arterial hypertension, stenocardia.[93]
It was shown that in children with pulmonary arterial hypertension due to congenital heart diseases the level of homocysteine is higher and the level of endogenous sulfur dioxide is lower than in normal control children. Moreover, these biochemical parameters strongly correlated to the severity of pulmonary arterial hypertension. Authors considered homocysteine to be one of useful biochemical markers of disease severity and sulfur dioxide metabolism to be one of potential therapeutic targets in those patients.[94]
Endogenous sulfur dioxide also has been shown to lower the proliferation rate of endothelial smooth muscle cells in blood vessels, via lowering the MAPK activity and activating adenylyl cyclase and protein kinase A.[95] Smooth muscle cell proliferation is one of important mechanisms of hypertensive remodeling of blood vessels and their stenosis, so it is an important pathogenetic mechanism in arterial hypertension and atherosclerosis.
Endogenous sulfur dioxide in low concentrations causes endothelium-dependent vasodilation. In higher concentrations it causes endothelium-independent vasodilation and has a negative inotropic effect on cardiac output function, thus effectively lowering blood pressure and myocardial oxygen consumption. The vasodilating effects of sulfur dioxide are mediated via ATP-dependent calcium channels and L-type ("dihydropyridine") calcium channels. Endogenous sulfur dioxide is also a potent antiinflammatory, antioxidant and cytoprotective agent. It lowers blood pressure and slows hypertensive remodeling of blood vessels, especially thickening of their intima. It also regulates lipid metabolism.[96]
Endogenous sulfur dioxide also diminishes myocardial damage, caused by isoproterenol adrenergic hyperstimulation, and strengthens the myocardial antioxidant defense reserve.[97]
Sulfur dioxide expelled from the gastrointestinal tract (through the process of flatulence) is thought to be one of the main constituents to the unpleasant smell colloquially referred to as a "fart".
Hydrogen cyanide
Some authors have shown that neurons can produce hydrogen cyanide upon activation of their opioid receptors by endogenous or exogenous opioids. They have also shown that neuronal production of HCN activates NMDA receptors and plays a role in signal transduction between neuronal cells (neurotransmission). Moreover, increased endogenous neuronal HCN production under opioids was seemingly needed for adequate opioid analgesia, as analgesic action of opioids was attenuated by HCN scavengers. They considered endogenous HCN to be a neuromodulator.[98]
It was also shown that, while stimulating muscarinic cholinergic receptors in cultured pheochromocytoma cells increases HCN production, in a living organism (in vivo) muscarinic cholinergic stimulation actually decreases HCN production.[99]
Leukocytes generate HCN during phagocytosis.[98]
The vasodilatation, caused by sodium nitroprusside, has been shown to be mediated not only by NO generation, but also by endogenous cyanide generation, which adds not only toxicity, but also some additional antihypertensive efficacy compared to nitroglycerine and other non-cyanogenic nitrates which do not cause blood cyanide levels to rise.[100]
Ammonia
Ammonia also plays a role in both normal and abnormal animal physiology. It is biosynthesised through normal amino acid metabolism and is toxic in high concentrations.[101] The liver converts ammonia to urea through a series of reactions known as the urea cycle. Liver dysfunction, such as that seen in cirrhosis, may lead to elevated amounts of ammonia in the blood (hyperammonemia). Likewise, defects in the enzymes responsible for the urea cycle, such as ornithine transcarbamylase, lead to hyperammonemia. Hyperammonemia contributes to the confusion and coma of hepatic encephalopathy, as well as the neurologic disease common in people with urea cycle defects and organic acidurias.[102]
Ammonia is important for normal animal acid/base balance. After formation of ammonium from glutamine, α-ketoglutarate may be degraded to produce two molecules of bicarbonate, which are then available as buffers for dietary acids. Ammonium is excreted in the urine, resulting in net acid loss. Ammonia may itself diffuse across the renal tubules, combine with a hydrogen ion, and thus allow for further acid excretion.[103]
Methane
Some authors have shown that endogenous methane is produced not only by the intestinal flora and then absorbed into the blood, but also is produced - in small amounts - by eukaryotic cells (during process of lipid peroxidation). And they have also shown that the endogenous methane production rises during an experimental mitochondrial hypoxia, for example, sodium azide intoxication. They thought that methane could be one of intercellular signals of hypoxia and stress.[104]
Other authors have shown that cellular methane production also rises during sepsis or bacterial endotoxemia, including an experimental imitation of endotoxemia by lipopolysaccharide (LPS) administration.[105]
Some other researchers have shown that methane, produced by the intestinal flora, is not fully "biologically neutral" to the intestine, and it participates in the normal physiologic regulation of peristalsis. And its excess causes not only belching, flatulence and belly pain, but also functional constipation.[106]
Ethylene
Ethylene serves as a hormone in plants.[107] It acts at trace levels throughout the life of the plant by stimulating or regulating the ripening of fruit, the opening of flowers, and the abscission (or shedding) of leaves. Commercial ripening rooms use "catalytic generators" to make ethylene gas from a liquid supply of ethanol. Typically, a gassing level of 500 to 2,000 ppm is used, for 24 to 48 hours. Care must be taken to control carbon dioxide levels in ripening rooms when gassing, as high temperature ripening (20 °C;68 °F) has been seen to produce CO2 levels of 10% in 24 hours.[108]
Ethylene has been used since the ancient Egyptians, who would gash figs in order to stimulate ripening (wounding stimulates ethylene production by plant tissues). The ancient Chinese would burn incense in closed rooms to enhance the ripening of pears. In 1864, it was discovered that gas leaks from street lights led to stunting of growth, twisting of plants, and abnormal thickening of stems.[107] In 1901, a Russian scientist named Dimitry Neljubow showed that the active component was ethylene.[109] Sarah Doubt discovered that ethylene stimulated abscission in 1917.[110] It wasn't until 1934 that Gane reported that plants synthesize ethylene.[111] In 1935, Crocker proposed that ethylene was the plant hormone responsible for fruit ripening as well as senescence of vegetative tissues.[112]
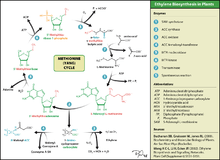
Ethylene is produced from essentially all parts of higher plants, including leaves, stems, roots, flowers, fruits, tubers, and seeds. Ethylene production is regulated by a variety of developmental and environmental factors. During the life of the plant, ethylene production is induced during certain stages of growth such as germination, ripening of fruits, abscission of leaves, and senescence of flowers. Ethylene production can also be induced by a variety of external aspects such as mechanical wounding, environmental stresses, and certain chemicals including auxin and other regulators.[113]
Ethylene is biosynthesized from the amino acid methionine to S-adenosyl-L-methionine (SAM, also called Adomet) by the enzyme Met Adenosyltransferase. SAM is then converted to 1-aminocyclopropane-1-carboxylic acid (ACC) by the enzyme ACC synthase (ACS). The activity of ACS determines the rate of ethylene production, therefore regulation of this enzyme is key for the ethylene biosynthesis. The final step requires oxygen and involves the action of the enzyme ACC-oxidase (ACO), formerly known as the ethylene forming enzyme (EFE). Ethylene biosynthesis can be induced by endogenous or exogenous ethylene. ACC synthesis increases with high levels of auxins, especially indole acetic acid (IAA) and cytokinins.
Ethylene is perceived by a family of five transmembrane protein dimers such as the ETR1 protein in Arabidopsis. The gene encoding an ethylene receptor has been cloned in Arabidopsis thaliana and then in tomato. Ethylene receptors are encoded by multiple genes in the Arabidopsis and tomato genomes. Mutations in any of the gene family, which comprises five receptors in Arabidopsis and at least six in tomato, can lead to insensitivity to ethylene.[114] DNA sequences for ethylene receptors have also been identified in many other plant species and an ethylene binding protein has even been identified in Cyanobacteria.[107]
Environmental cues such as flooding, drought, chilling, wounding, and pathogen attack can induce ethylene formation in plants. In flooding, roots suffer from lack of oxygen, or anoxia, which leads to the synthesis of 1-aminocyclopropane-1-carboxylic acid (ACC). ACC is transported upwards in the plant and then oxidized in leaves. The ethylene produced causes nastic movements (epinasty) of the leaves, perhaps helping the plant to lose water.[115]
Ethylene in plant induces such responses:
- Seedling triple response, thickening and shortening of hypocotyl with pronounced apical hook.
- In pollination, when the pollen reaches the stigma, the precursor of the ethene, ACC, is secreted to the petal, the ACC releases ethylene with ACC oxidase.
- Stimulates leaf and flower senescence
- Stimulates senescence of mature xylem cells in preparation for plant use
- Induces leaf abscission
- Induces seed germination
- Induces root hair growth — increasing the efficiency of water and mineral absorption
- Induces the growth of adventitious roots during flooding
- Stimulates epinasty — leaf petiole grows out, leaf hangs down and curls into itself
- Stimulates fruit ripening
- Induces a climacteric rise in respiration in some fruit which causes a release of additional ethylene.
- Affects gravitropism
- Stimulates nutational bending
- Inhibits stem growth and stimulates stem and cell broadening and lateral branch growth outside of seedling stage (see Hyponastic response)
- Interference with auxin transport (with high auxin concentrations)
- Inhibits shoot growth and stomatal closing except in some water plants or habitually flooded ones such as some rice varieties, where the opposite occurs (conserving CO
2 and O
2) - Induces flowering in pineapples
- Inhibits short day induced flower initiation in Pharbitus nil[116] and Chrysanthemum morifolium[117]
Small amounts of endogenous ethylene are also produced in mammals, including humans, due to lipid peroxidation. Some of endogenous ethylene is then oxidized to ethylene oxide, which is able to alkylate DNA and proteins, including hemoglobin (forming a specific adduct with its N-terminal valine, N-hydroxyethyl-valine).[118] Endogenous ethylene oxide, just as like environmental (exogenous) one, can alkylate guanine in DNA, forming an adduct 7-(2-hydroxyethyl)-guanine, and this poses an intrinsic carcinogenic risk.[119] It is also mutagenic.[120][121]
References
- Mustafa AK, Gadalla MM, Snyder SH (2009). "Signaling by gasotransmitters". Sci Signal. 2 (68): re2. doi:10.1126/scisignal.268re2. PMC 2744355. PMID 19401594.
- Gillman MA, Lichtigfeld FJ (January 1981). "A comparison of the effects of morphine sulphate and nitrous oxide analgesia on chronic pain states in man". J. Neurol. Sci. 49 (1): 41–45. doi:10.1016/0022-510X(81)90186-6. PMID 7205318.
- Gillman MA, Lichtigfeld FJ (February 1981). "The similarity of the action of nitrous oxide and morphine". Pain. 10 (1): 110. doi:10.1016/0304-3959(81)90054-3. PMID 7232008.
- Gillman MA, Lichtigfeld FJ (May 1983). "Nitrous oxide interacts with opioid receptors: more evidence". Anesthesiology. 58 (5): 483–4. doi:10.1097/00000542-198305000-00021. PMID 6301312.
- Daras, C; Cantrill, R; Gillman, MA (1983). "(3H)Naloxone displacement: evidence for nitrous oxide as opioid receptor agonist". Eur J Pharmacol. 89 (1–2): 177–178. doi:10.1016/0014-2999(83)90626-x.
- Ori, C.; Ford-Rice, F; London, E.D. (1989). "Effects of nitrous oxide and halothane on mu and kappa opioid receptors in guinea-pig brain". Anesthesiology. 70 (3): 541–544. doi:10.1097/00000542-198903000-00027.
- Wang, R (2002). "Two's company, three's a crowd - Can H
2S be the third endogenous gaseous transmitter?". FASEB Journal. 16 (13): 1792–1798. doi:10.1096/fj.02-0211hyp. PMID 12409322. - Wang R (ed) (2004) Signal Transduction and the Gasotransmitters: NO, CO and H2S in Biology and Medicine. Humana Press, New Jersey, USA.
- Weller, Richard, Could the Sun be good for your heart? TedxGlasgow. Filmed March 2012, posted January 2013
- Roszer, T (2012) The Biology of Subcellular Nitric Oxide. ISBN 978-94-007-2818-9
- Stryer, Lubert (1995). Biochemistry, 4th Edition. W.H. Freeman and Company. p. 732. ISBN 978-0-7167-2009-6.
- "Plant-based Diets | Plant-based Foods | Beetroot Juice | Nitric Oxide Vegetables". Berkeley Test. Retrieved 2013-10-04.
- Ghosh, S. M.; Kapil, V.; Fuentes-Calvo, I.; Bubb, K. J.; Pearl, V.; Milsom, A. B.; Khambata, R.; Maleki-Toyserkani, S.; Yousuf, M.; Benjamin, N.; Webb, A. J.; Caulfield, M. J.; Hobbs, A. J.; Ahluwalia, A. (2013). "Enhanced Vasodilator Activity of Nitrite in Hypertension: Critical Role for Erythrocytic Xanthine Oxidoreductase and Translational Potential". Hypertension. 61 (5): 1091–102. doi:10.1161/HYPERTENSIONAHA.111.00933. PMID 23589565.
- Webb, A. J.; Patel, N.; Loukogeorgakis, S.; Okorie, M.; Aboud, Z.; Misra, S.; Rashid, R.; Miall, P.; Deanfield, J.; Benjamin, N.; MacAllister, R.; Hobbs, A. J.; Ahluwalia, A. (2008). "Acute Blood Pressure Lowering, Vasoprotective, and Antiplatelet Properties of Dietary Nitrate via Bioconversion to Nitrite". Hypertension. 51 (3): 784–90. doi:10.1161/HYPERTENSIONAHA.107.103523. PMC 2839282. PMID 18250365.
- Hezel, MP; Weitzberg, E (2013). "The oral microbiome and nitric oxide homoeostasis". Oral Diseases. 21 (1): 7–16. doi:10.1111/odi.12157. PMID 23837897.
- Green, Shawn J. (2013-07-25). "Turning DASH Strategy into Reality for Improved Cardio Wellness Outcomes: Part II". Real World Health Care. Retrieved 2013-10-04.
- Proctor, PH (August 1989). "Endothelium-Derived Relaxing Factor and Minoxidil: Active Mechanisms in Hair Growth". Archives of Dermatology. 125 (8): 1146. doi:10.1001/archderm.1989.01670200122026. PMID 2757417.
- Dessy, C.; Ferron, O. (2004). "Pathophysiological Roles of Nitric Oxide: In the Heart and the Coronary Vasculature". Current Medical Chemistry – Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry. 3 (3): 207–216. doi:10.2174/1568014043355348.
- Osanai, T; Fujiwara, N; Saitoh, M; Sasaki, S; Tomita, H; Nakamura, M; Osawa, H; Yamabe, H; Okumura, K (2002). "Relationship between salt intake, nitric oxide, and asymmetric dimethylarginine and its relevance to patients with end-stage renal disease". Blood Purification. 20 (5): 466–8. doi:10.1159/000063555. PMID 12207094.
- Green, SJ; Mellouk, S; Hoffman, SL; Meltzer, MS; Nacy, CA (1990). "Cellular mechanisms of nonspecific immunity to intracellular infection: Cytokine-induced synthesis of toxic nitrogen oxides from L-arginine by macrophages and hepatocytes". Immunology Letters. 25 (1–3): 15–9. doi:10.1016/0165-2478(90)90083-3. PMID 2126524.
- Gorczyniski and Stanely, Clinical Immunology. Landes Bioscience; Austin, TX. ISBN 1-57059-625-5
- Green, SJ; Nacy, CA; Schreiber, RD; Granger, DL; Crawford, RM; Meltzer, MS; Fortier, AH (1993). "Neutralization of gamma interferon and tumor necrosis factor alpha blocks in vivo synthesis of nitrogen oxides from L-arginine and protection against Francisella tularensis infection in Mycobacterium bovis BCG-treated mice". Infection and Immunity. 61 (2): 689–98. PMC 302781. PMID 8423095.
- Kamijo, R; Gerecitano, J; Shapiro, D; Green, SJ; Aguet, M; Le, J; Vilcek, J (1995). "Generation of nitric oxide and clearance of interferon-gamma after BCG infection are impaired in mice that lack the interferon-gamma receptor". Journal of Inflammation. 46 (1): 23–31. PMID 8832969.
- Green, SJ; Scheller, LF; Marletta, MA; Seguin, MC; Klotz, FW; Slayter, M; Nelson, BJ; Nacy, CA (1994). "Nitric oxide: Cytokine-regulation of nitric oxide in host resistance to intracellular pathogens" (PDF). Immunology Letters. 43 (1–2): 87–94. doi:10.1016/0165-2478(94)00158-8. hdl:2027.42/31140. PMID 7537721.
- Green, SJ; Crawford, RM; Hockmeyer, JT; Meltzer, MS; Nacy, CA (1990). "Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-gamma-stimulated macrophages by induction of tumor necrosis factor-alpha". Journal of Immunology. 145 (12): 4290–7. PMID 2124240.
- Seguin, M. C.; Klotz, FW; Schneider, I; Weir, JP; Goodbary, M; Slayter, M; Raney, JJ; Aniagolu, JU; Green, SJ (1994). "Induction of nitric oxide synthase protects against malaria in mice exposed to irradiated Plasmodium berghei infected mosquitoes: Involvement of interferon gamma and CD8+ T cells". Journal of Experimental Medicine. 180 (1): 353–8. doi:10.1084/jem.180.1.353. PMC 2191552. PMID 7516412.
- Mellouk, S; Green, SJ; Nacy, CA; Hoffman, SL (1991). "IFN-gamma inhibits development of Plasmodium berghei exoerythrocytic stages in hepatocytes by an L-arginine-dependent effector mechanism". Journal of Immunology. 146 (11): 3971–6. PMID 1903415.
- Klotz, FW; Scheller, LF; Seguin, MC; Kumar, N; Marletta, MA; Green, SJ; Azad, AF (1995). "Co-localization of inducible-nitric oxide synthase and Plasmodium berghei in hepatocytes from rats immunized with irradiated sporozoites". Journal of Immunology. 154 (7): 3391–5. PMID 7534796.
- Wink, D.; Kasprzak, K.; Maragos, C.; Elespuru, R.; Misra, M; Dunams, T.; Cebula, T.; Koch, W.; Andrews, A.; Allen, J.; Et, al. (1991). "DNA deaminating ability and genotoxicity of nitric oxide and its progenitors". Science. 254 (5034): 1001–3. Bibcode:1991Sci...254.1001W. doi:10.1126/science.1948068. PMID 1948068.
- Nguyen, T.; Brunson, D.; Crespi, C. L.; Penman, B. W.; Wishnok, J. S.; Tannenbaum, S. R. (1992). "DNA Damage and Mutation in Human Cells Exposed to Nitric Oxide in vitro". Proceedings of the National Academy of Sciences. 89 (7): 3030–3034. Bibcode:1992PNAS...89.3030N. doi:10.1073/pnas.89.7.3030. PMC 48797. PMID 1557408. Free text.
- Li, Chun-Qi; Pang, Bo; Kiziltepe, Tanyel; Trudel, Laura J.; Engelward, Bevin P.; Dedon, Peter C.; Wogan, Gerald N. (2006). "Threshold Effects of Nitric Oxide-Induced Toxicity and Cellular Responses in Wild-Type and p53-Null Human Lymphoblastoid Cells". Chemical Research in Toxicology. 19 (3): 399–406. doi:10.1021/tx050283e. PMC 2570754. PMID 16544944. free text
- Hibbs, John B.; Taintor, Read R.; Vavrin, Zdenek; Rachlin, Elliot M. (1988). "Nitric oxide: A cytotoxic activated macrophage effector molecule". Biochemical and Biophysical Research Communications. 157 (1): 87–94. doi:10.1016/S0006-291X(88)80015-9. PMID 3196352.
- Janeway, C. A.; et al. (2005). Immunobiology: the immune system in health and disease (6th ed.). New York: Garland Science. ISBN 978-0-8153-4101-7.
- Jacobs, Lotte; Nawrot, Tim S; De Geus, Bas; Meeusen, Romain; Degraeuwe, Bart; Bernard, Alfred; Sughis, Muhammad; Nemery, Benoit; Panis, Luc (2010). "Subclinical responses in healthy cyclists briefly exposed to traffic-related air pollution: An intervention study". Environmental Health. 9: 64. doi:10.1186/1476-069X-9-64. PMC 2984475. PMID 20973949.
- Corpas, F. J.; Barroso, JB; Carreras, A; Quirós, M; León, AM; Romero-Puertas, MC; Esteban, FJ; Valderrama, R; Palma, JM; Sandalio, LM; Gómez, M; Del Río, LA (2004). "Cellular and subcellular localization of endogenous nitric oxide in young and senescent pea plants". Plant Physiology. 136 (1): 2722–33. doi:10.1104/pp.104.042812. PMC 523336. PMID 15347796.
- Corpas, F. J.; Barroso, Juan B.; Carreras, Alfonso; Valderrama, Raquel; Palma, José M.; León, Ana M.; Sandalio, Luisa M.; Del Río, Luis A (2006). "Constitutive arginine-dependent nitric oxide synthase activity in different organs of pea seedlings during plant development". Planta. 224 (2): 246–54. doi:10.1007/s00425-005-0205-9. PMID 16397797.
- Valderrama, R.; Corpas, Francisco J.; Carreras, Alfonso; Fernández-Ocaña, Ana; Chaki, Mounira; Luque, Francisco; Gómez-Rodríguez, María V.; Colmenero-Varea, Pilar; Del Río, Luis A.; Barroso, Juan B. (2007). "Nitrosative stress in plants". FEBS Lett. 581 (3): 453–61. doi:10.1016/j.febslet.2007.01.006. PMID 17240373.
- Corpas, F. J.; Barroso, Juan B.; Del Rio, Luis A. (2004). "Enzymatic sources of nitric oxide in plant cells – beyond one protein–one function". New Phytologist. 162 (2): 246–7. doi:10.1111/j.1469-8137.2004.01058.x.
- Siegel-Itzkovich, J. (1999). "Viagra makes flowers stand up straight". BMJ. 319 (7205): 274. doi:10.1136/bmj.319.7205.274a. PMC 1126920. PMID 10426722.
- van Faassen, E. and Vanin, A. (eds.) (2007) Radicals for life: The various forms of nitric oxide. Elsevier, Amsterdam, ISBN 978-0-444-52236-8
- van Faassen, E. and Vanin, A. (2004) "Nitric Oxide", in Encyclopedia of Analytical Science, 2nd ed., Elsevier, ISBN 0127641009.
- Shami, PJ; Moore, JO; Gockerman, JP; Hathorn, JW; Misukonis, MA; Weinberg, JB (1995). "Nitric oxide modulation of the growth and differentiation of freshly isolated acute non-lymphocytic leukemia cells". Leukemia Research. 19 (8): 527–33. doi:10.1016/0145-2126(95)00013-E. PMID 7658698.
- Kaibori M.; Sakitani K.; Oda M.; Kamiyama Y.; Masu Y.; Okumura T. (1999). "Immunosuppressant FK506 inhibits inducible nitric oxide synthase gene expression at a step of NF-κB activation in rat hepatocytes". J. Hepatol. 30 (6): 1138–1145. doi:10.1016/S0168-8278(99)80270-0. PMID 10406194.
- Rhoades, RA; Tanner, GA (2003). "Medical physiology 2nd edition". Comparative Biochemistry and Physiology. A, Comparative Physiology. 53 (1): 105–7. PMID 174.
- Hogg N, Singh RJ, Kalyanaraman B (March 18, 1996). "The role of glutathione in the transport and catabolism of nitric oxide". FEBS Letters. 382 (3): 223–228. doi:10.1016/0014-5793(96)00086-5. PMID 8605974.
- DeMaster EG, Quast BJ, Redfern B, Nagasawa HT (Sep 12, 1995). "Reaction of nitric oxide with the free sulfhydryl group of human serum albumin yields a sulfenic acid and nitrous oxide". Biochemistry. 34 (36): 11494–11499. doi:10.1021/bi00036a023. PMID 7547878.
- Hyun J, Chaudhuri G, Fukuto JM (September 1, 1999). "The Reductive Metabolism of Nitric Oxide in Hepatocytes: Possible Interaction with Thiols". Drug Metabolism and Disposition. 27 (9): 1005–1009. PMID 10460799.
- Finck, A. D., Samaniego, E., Ngai, S.H. [1995]. Nitrous oxide selectively releases met5-enkephalin and met5-enkephalin-arg6-phe7 into canine third ventricular cerebrospinal fluid. Anesthesia and Analgesia 80: 664-70
- Jevtović-Todorović V, Todorović SM, Mennerick S, Powell S, Dikranian K, Benshoff N, Zorumski CF, Olney JW (Apr 1998). "Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin". Nat Med. 4 (4): 460–463. doi:10.1038/nm0498-460. PMID 9546794.
- Christensen B, Refsum H, Garras A, Ueland PM (Jun 1992). "Homocysteine remethylation during nitrous oxide exposure of cells cultured in media containing various concentrations of folates". J Pharmacol Exp Ther. 261 (3): 1096–1105. PMID 1602376.
- Koblin DD, Waskell L, Watson JE, Stokstad EL, Eger EI 2nd (Feb 1982). "Nitrous oxide inactivates methionine synthetase in human liver". Anesth Analg. 61 (2): 75–78. doi:10.1213/00000539-198202000-00001. PMID 7198880.
- Sampath V, Zhao XJ, Caughey WS (Apr 27, 2001). "Anesthetic-like interactions of nitric oxide with albumin and hemeproteins. A mechanism for control of protein function". The Journal of Biological Chemistry. 276 (17): 13635–13643. doi:10.1074/jbc.M006588200. PMID 11278308.
- Dong A, Huang P, Zhao XJ, Sampath V, Caughey WS (September 30, 1994). "Characterization of sites occupied by the anesthetic nitrous oxide within proteins by infrared spectroscopy". The Journal of Biological Chemistry. 269 (39): 23911–23917. PMID 7929038.
- Einarsdóttir O, Caughey WS (5 Jul 1988). "Interactions of the anesthetic nitrous oxide with bovine heart cytochrome c oxidase. Effects on protein structure, oxidase activity, and other properties". The Journal of Biological Chemistry. 263 (19): 9199–9205. PMID 2837481.
- Gillman MA, Lichtigfeld FJ (March 1985). "Nitrous oxide acts directly at the mu opioid receptor". Anesthesiology. 62 (3): 375–376. doi:10.1097/00000542-198503000-00040. PMID 2983587.
- Wu, L; Wang, R (December 2005). "Carbon Monoxide: Endogenous Production, Physiological Functions, and Pharmacological Applications". Pharmacol Rev. 57 (4): 585–630. doi:10.1124/pr.57.4.3. PMID 16382109.
- Olas, Beata (25 April 2014). "Carbon monoxide is not always a poison gas for human organism: Physiological and pharmacological features of CO". Chemico-Biological Interactions. 222 (5 October 2014): 37–43. doi:10.1016/j.cbi.2014.08.005. PMID 25168849.
- Verma, A; Hirsch, D.; Glatt, C.; Ronnett, G.; Snyder, S. (1993). "Carbon monoxide: A putative neural messenger". Science. 259 (5093): 381–4. Bibcode:1993Sci...259..381V. doi:10.1126/science.7678352. PMID 7678352.
- Kolata, Gina (January 26, 1993). "Carbon Monoxide Gas Is Used by Brain Cells As a Neurotransmitter". The New York Times. Retrieved May 2, 2010.
- Li, L; Hsu, A; Moore, PK (2009). "Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation—a tale of three gases!". Pharmacology & Therapeutics. 123 (3): 386–400. doi:10.1016/j.pharmthera.2009.05.005. PMID 19486912.
- Johnson, Carolyn Y. (October 16, 2009). "Poison gas may carry a medical benefit". The Boston Globe. Retrieved October 16, 2009.
- Kerek F (Sep 2000). "The structure of the digitalislike and natriuretic factors identified as macrocyclic derivatives of the inorganic carbon suboxide". Hypertension Research. 23 (Suppl S33): S33–38. doi:10.1291/hypres.23.Supplement_S33. PMID 11016817.
- Stimac R, Kerek F, Apell HJ (Apr 2003). "Macrocyclic carbon suboxide oligomers as potent inhibitors of the Na,K-ATPase". Annals of the New York Academy of Sciences. 986 (1): 327–329. Bibcode:2003NYASA.986..327S. doi:10.1111/j.1749-6632.2003.tb07204.x. PMID 12763840.
- Kerek F, Stimac R, Apell HJ, Freudenmann F, Moroder L (23 December 2002). "Characterization of the macrocyclic carbon suboxide factors as potent Na,K-ATPase and SR Ca-ATPase inhibitors". Biochimica et Biophysica Acta (BBA) - Biomembranes. 1567 (1–2): 213–220. doi:10.1016/S0005-2736(02)00609-0. PMID 12488055.
- Tubaro E. (Jun 1966). "Carbon suboxide, the probable precursor of an antitumor cellular sustance [sic]: retina". Boll Chim Farm (in Italian). 105 (6): 415–416. PMID 6005012.
- Lefer, David J. (November 2007). "A new gaseous signaling molecule emerges: Cardioprotective role of hydrogen sulfide". PNAS. 104 (46): 17907–17908. Bibcode:2007PNAS..10417907L. doi:10.1073/pnas.0709010104. PMC 2084269. PMID 17991773.
- Kimura, Hideo (2002). "Hydrogen sulfide as a neuromodulator". Molecular Neurobiology. 26 (1): 13–19. doi:10.1385/MN:26:1:013. PMID 12392053.
- Kamoun, Pierre (July 2004). "H2S, a new neuromodulator". Médecine/Sciences. 20 (6–7): 697–700. doi:10.1051/medsci/2004206-7697. PMID 15329822.
- Benavides, Gloria A; Squadrito, Giuseppe L; Mills, Robert W; Patel, Hetal D; Isbell, T Scott; Patel, Rakesh P; Darley-Usmar, Victor M; Doeller, Jeannette E; Kraus, David W (2007-11-13). "Hydrogen sulfide mediates the vasoactivity of garlic". Proceedings of the National Academy of Sciences of the United States of America. 104 (46): 17977–17982. Bibcode:2007PNAS..10417977B. doi:10.1073/pnas.0705710104. PMC 2084282. PMID 17951430.
- "Toxic Gas, Lifesaver", Scientific American, March 2010
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I, Martin E, Szabo C (2012). "Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation". Proc. Natl. Acad. Sci. U.S.A. 109 (23): 9161–6. Bibcode:2012PNAS..109.9161C. doi:10.1073/pnas.1202916109. PMC 3384190. PMID 22570497.
- Filipovic, M. R.; Miljkovic, J. L.; Nauser, T.; Royzen, M.; Klos, K.; Shubina, T.; Koppenol, W. H.; Lippard, S. J.; Ivanović-Burmazović, I. (2012). "Chemical Characterization of the SmallestS-Nitrosothiol, HSNO; Cellular Cross-talk of H2S andS-Nitrosothiols". Journal of the American Chemical Society. 134 (29): 12016–12027. doi:10.1021/ja3009693. PMC 3408084. PMID 22741609.
- d'Emmanuele di Villa Bianca R, Sorrentino R, Maffia P, Mirone V, Imbimbo C, Fusco F, De Palma R, Ignarro LJ, Cirino G (2009). "Hydrogen sulfide as a mediator of human corpus cavernosum smooth-muscle relaxation". PNAS. 106 (11): 4513–8. Bibcode:2009PNAS..106.4513D. doi:10.1073/pnas.0807974105. PMC 2657379. PMID 19255435.
- "Hydrogen Sulfide: Potential Help for ED". WebMD. March 2, 2009.
- King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ (January 2014). "Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent". PNAS. 111 (Early Edition): 3182–3187. Bibcode:2014PNAS..111.3182K. doi:10.1073/pnas.1321871111. PMC 3939925. PMID 24516168.
- Alp, Nicholas; Channon (2003). "Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease". Journal of the American Heart Association. 24 (3): 413–420. doi:10.1161/01.atv.0000110785.96039.f6. PMID 14656731.
- Boerth NJ, Dey NB, Cornwell TL, Lincoln TM (1997). "Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype". Journal of Vascular Research. 34 (4): 245–259. doi:10.1159/000159231. PMID 9256084.
- Lincoln, T. M.; Cornwell, Taylor (March 1990). "cGMP-dependent protein kinase mediates the reduction of Ca2+ by cAMP in vascular smooth muscle cells". American Journal of Physiology. 258 (3): C399–C407. doi:10.1152/ajpcell.1990.258.3.C399. PMID 2156436.
- Eto, Ko; Takashi Asada; Kunimasa Arima; Takao Makifuchi; Hideo Kimura (2002-05-24). "Brain hydrogen sulfide is severely decreased in Alzheimer's disease". Biochemical and Biophysical Research Communications. 293 (5): 1485–1488. doi:10.1016/S0006-291X(02)00422-9. PMID 12054683.
- Hu, L. F.; Lu, M.; Tiong, C. X.; Dawe, G. S.; Hu, G.; Bian, J. S. (2010). "Neuroprotective effects of hydrogen sulfide on Parkinson's disease rat models". Aging Cell. 9 (2): 135–146. doi:10.1111/j.1474-9726.2009.00543.x. PMID 20041858.
- Mice put in 'suspended animation', BBC News, 21 April 2005
- Gas induces 'suspended animation', BBC News, 9 October 2006
- Florian B, Vintilescu R, Balseanu AT, Buga AM, Grisk O, Walker LC, Kessler C, Popa-Wagner A (2008). "Long-term hypothermia reduces infarct volume in aged rats after focal ischemia". Neurosci. Lett. 438 (2): 180–5. doi:10.1016/j.neulet.2008.04.020. PMID 18456407.
- Mark B. Roth and Todd Nystul. Buying Time in Suspended Animation. Scientific American, 1 June 2005
- Li, Jia; Zhang, Gencheng; Cai, Sally; Redington, Andrew N (January 2008). "Effect of inhaled hydrogen sulfide on metabolic responses in anesthetized, paralyzed, and mechanically ventilated piglets". Pediatric Critical Care Medicine. 9 (1): 110–112. doi:10.1097/01.PCC.0000298639.08519.0C. PMID 18477923. Retrieved 2008-02-07.
H2S does not appear to have hypometabolic effects in ambiently cooled large mammals and conversely appears to act as a hemodynamic and metabolic stimulant.
- Haouzi P, Notet V, Chenuel B, Chalon B, Sponne I, Ogier V, et al. (2008). "H2S induced hypometabolism in mice is missing in sedated sheep". Respir Physiol Neurobiol. 160 (1): 109–15. doi:10.1016/j.resp.2007.09.001. PMID 17980679.
- "Mark Roth: Suspended animation is within our grasp".
- "IK-1001 (Sodium Sulfide (Na2S) for Injection) in Subjects With Acute ST-Segment Elevation Myocardial Infarction". ClinicalTrials.gov. 2010-11-04.
This study has been withdrawn prior to enrollment. ( Company decision. Non-safety related )
- "Reduction of Ischemia-Reperfusion Mediated Cardiac Injury in Subjects Undergoing Coronary Artery Bypass Graft Surgery". ClinicalTrials.gov. 2011-08-03.
This study has been terminated. ( Study Terminated - Company decision )
- Liu, D.; Jin, H; Tang, C; Du, J (2010). "Sulfur dioxide: a novel gaseous signal in the regulation of cardiovascular functions". Mini-Reviews in Medicinal Chemistry. 10 (11): 1039–1045. doi:10.2174/1389557511009011039. PMID 20540708.
- Chen S, Zheng S, Liu Z, Tang C, Zhao B, Du J, Jin H (Feb 2015). "Endogenous sulfur dioxide protects against oleic acid-induced acute lung injury in association with inhibition of oxidative stress in rats". Lab. Invest. 95 (2): 142–156. doi:10.1038/labinvest.2014.147. PMID 25581610.
- Tian H. (Nov 2014). "Advances in the study on endogenous sulfur dioxide in the cardiovascular system". Chin Med J. 127 (21): 3803–3807. PMID 25382339.
- Yang R, Yang Y, Dong X, Wu X, Wei Y (Aug 2014). "Correlation between endogenous sulfur dioxide and homocysteine in children with pulmonary arterial hypertension associated with congenital heart disease". Zhonghua Er Ke Za Zhi (in Chinese). 52 (8): 625–629. PMID 25224243.
- Liu D, Huang Y, Bu D, Liu AD, Holmberg L, Jia Y, Tang C, Du J, Jin H (May 2014). "Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the Erk/MAP kinase pathway mediated by cAMP/PKA signaling". Cell Death Dis. 5 (5): e1251. doi:10.1038/cddis.2014.229. PMC 4047873. PMID 24853429.
- Wang XB, Jin HF, Tang CS, Du JB (16 Nov 2011). "The biological effect of endogenous sulfur dioxide in the cardiovascular system". Eur J Pharmacol. 670 (1): 1–6. doi:10.1016/j.ejphar.2011.08.031. PMID 21925165.
- Liang Y, Liu D, Ochs T, Tang C, Chen S, Zhang S, Geng B, Jin H, Du J (Jan 2011). "Endogenous sulfur dioxide protects against isoproterenol-induced myocardial injury and increases myocardial antioxidant capacity in rats". Lab. Invest. 91 (1): 12–23. doi:10.1038/labinvest.2010.156. PMID 20733562.
- Borowitz JL, Gunasekar PG, Isom GE (12 Sep 1997). "Hydrogen cyanide generation by mu-opiate receptor activation: possible neuromodulatory role of endogenous cyanide". Brain Res. 768 (1–2): 294–300. doi:10.1016/S0006-8993(97)00659-8. PMID 9369328.
- Gunasekar PG, Prabhakaran K, Li L, Zhang L, Isom GE, Borowitz JL (May 2004). "Receptor mechanisms mediating cyanide generation in PC12 cells and rat brain". Neurosci Res. 49 (1): 13–18. doi:10.1016/j.neures.2004.01.006. PMID 15099699.
- Smith RP, Kruszyna H (Jan 1976). "Toxicology of some inorganic antihypertensive anions". Fed Proc. 35 (1): 69–72. PMID 1245233.
- "PubChem Substance Summary". Retrieved 7 July 2009.
- Zschocke, Johannes; Georg Hoffman (2004). Vademecum Metabolism. Friedrichsdorf, Germany: Milupa GmbH.
- Rose, Burton; Helmut Rennke (1994). Renal Pathophysiology. Baltimore: Williams & Wilkins. ISBN 978-0-683-07354-6.
- Eszter Tuboly; Andrea Szabó; Dénes Garab; Gábor Bartha; Ágnes Janovszky; Gábor Ero″s; Anna Szabó; Árpád Mohácsi; Gábor Szabó; József Kaszaki; Miklós Ghyczy; Mihály Boros (15 January 2013). "Methane biogenesis during sodium azide-induced chemical hypoxia in rats". American Journal of Physiology. Cell Physiology. 304 (2): 207–214. doi:10.1152/ajpcell.00300.2012. PMID 23174561.
- Tuboly E, Szabó A, Erős G, Mohácsi A, Szabó G, Tengölics R, Rákhely G, Boros M (Dec 2013). "Determination of endogenous methane formation by photoacoustic spectroscopy" (PDF). J Breath Res. 7 (4): 046004. Bibcode:2013JBR.....7d6004T. doi:10.1088/1752-7155/7/4/046004. PMID 24185326.
- Sahakian AB, Jee SR, Pimentel M (Aug 2010). "Methane and the gastrointestinal tract". Dig Dis Sci. 55 (8): 2135–2143. doi:10.1007/s10620-009-1012-0. PMID 19830557.
- Lin, Z.; Zhong, S.; Grierson, D. (2009). "Recent advances in ethylene research". J. Exp. Bot. 60 (12): 3311–36. doi:10.1093/jxb/erp204. PMID 19567479.
- External Link to More on Ethylene Gassing and Carbon Dioxide Control. ne-postharvest.com
- Neljubov D. (1901). "Uber die horizontale Nutation der Stengel von Pisum sativum und einiger anderen Pflanzen". Beih Bot Zentralbl. 10: 128–139.
- Doubt, Sarah L. (1917). "The Response of Plants to Illuminating Gas". Botanical Gazette. 63 (3): 209–224. doi:10.1086/332006. hdl:2027/mdp.39015068299380. JSTOR 2469142.
- Gane R. (1934). "Production of ethylene by some fruits". Nature. 134 (3400): 1008. Bibcode:1934Natur.134.1008G. doi:10.1038/1341008a0.
- Crocker W, Hitchcock AE, Zimmerman PW. (1935) "Similarities in the effects of ethlyene and the plant auxins". Contrib. Boyce Thompson Inst. 7. 231-48. Auxins Cytokinins IAA Growth substances, Ethylene
- Yang, S. F.; Hoffman N. E. (1984). "Ethylene biosynthesis and its regulation in higher plants". Annu. Rev. Plant Physiol. 35: 155–89. doi:10.1146/annurev.pp.35.060184.001103.
- Bleecker, A. B.; Esch, J. J.; Hall, A. E.; Rodríguez, F. I.; Binder, B. M. (1998). "The ethylene-receptor family from Arabidopsis: Structure and function". Philosophical Transactions of the Royal Society B: Biological Sciences. 353 (1374): 1405–12. doi:10.1098/rstb.1998.0295. PMC 1692356. PMID 9800203.
- Explaining Epinasty. planthormones.inf
- Wilmowicz E, Kesy J, Kopcewicz J (December 2008). "Ethylene and ABA interactions in the regulation of flower induction in Pharbitis nil". J. Plant Physiol. 165 (18): 1917–28. doi:10.1016/j.jplph.2008.04.009. PMID 18565620.
- Cockshull KE, Horridge JS (1978). "2-Chloroethylphosphonic Acid and Flower Initiation by Chrysanthemum morifolium Ramat. In Short Days and in Long Days". Journal of Horticultural Science & Biotechnology. 53 (2): 85–90. doi:10.1080/00221589.1978.11514799.
- Filser JG, Denk B, Törnqvist M, Kessler W, Ehrenberg L (1992). "Pharmacokinetics of ethylene in man; body burden with ethylene oxide and hydroxyethylation of hemoglobin due to endogenous and environmental ethylene". Arch. Toxicol. 66 (3): 157–163. doi:10.1007/bf01974008. PMID 1303633.
- Bolt HM, Leutbecher M, Golka K (1997). "A note on the physiological background of the ethylene oxide adduct 7-(2-hydroxyethyl)guanine in DNA from human blood". Arch. Toxicol. 71 (11): 719–721. doi:10.1007/s002040050451. PMID 9363847.
- Csanády GA, Denk B, Pütz C, Kreuzer PE, Kessler W, Baur C, Gargas ML, Filser JG (May 15, 2000). "A physiological toxicokinetic model for exogenous and endogenous ethylene and ethylene oxide in rat, mouse, and human: formation of 2-hydroxyethyl adducts with hemoglobin and DNA". Toxicol Appl Pharmacol. 165 (1): 1–26. doi:10.1006/taap.2000.8918. PMID 10814549.
- Thier R, Bolt HM (Sep 2000). "Carcinogenicity and genotoxicity of ethylene oxide: new aspects and recent advances". Crit Rev Toxicol. 30 (5): 595–608. doi:10.1080/10408440008951121. PMID 11055837.
External links
