Dermatopathia pigmentosa reticularis
Dermatopathia pigmentosa reticularis is a rare, autosomal dominant[2] congenital disorder that is a form of ectodermal dysplasia. Dermatopathia pigmentosa reticularis is composed of the triad of generalized reticulate hyperpigmentation, noncicatricial alopecia, and onychodystrophy.[3]:856
| Dermatopathia pigmentosa reticularis | |
|---|---|
| Other names | Dermatopathic pigmentosa reticularis[1]:511 |
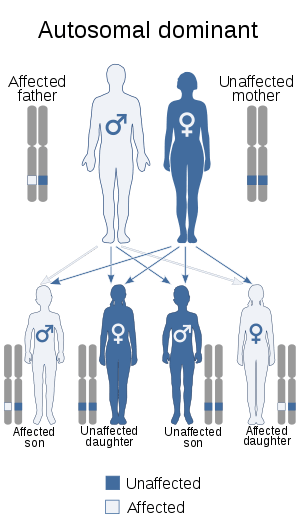 | |
| Dermatopathia pigmentosa reticularis has an autosomal dominant pattern of inheritance | |
| Specialty | Medical genetics |
Presentation
Symptoms include lack of sweat glands, thin hair, brittle nails, mottled skin, and lack of fingerprints.[4]
DPR is very similar to the related Naegeli-Franceschetti-Jadassohn syndrome. Both cause an affected person to lack fingerprints, have a lace-like pattern of hyperpigmentation and hyperkeratosis of the palms of the hands and soles of the feet. DPR is distinguished from NFJS by the duration of hyperpigmentation and lack of dental abnormalities.[5]
Cause
DPR is caused by a mutation in the keratin 14 gene.[6]
Treatment
References
- Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- Heimer WL II, Brauner G, James WD (1992). "Dermatopathia pigmentosa reticularis: a report of a family demonstrating autosomal dominant inheritance". J Am Acad Dermatol. 26 (2 pt. 2): 298–301. doi:10.1016/0190-9622(92)70039-I. PMID 1303619.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- "OMIM Clinical Synopsis - #125595 - DERMATOPATHIA PIGMENTOSA RETICULARIS; DPR". www.omim.org. Retrieved 13 October 2018.
- "OMIM Entry - # 125595 - DERMATOPATHIA PIGMENTOSA RETICULARIS; DPR". www.omim.org. Retrieved 13 October 2018.
- Lugassy J, Itin P, Ishida-Yamamoto A, et al. (October 2006). "Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis: two allelic ectodermal dysplasias caused by dominant mutations in KRT14". Am. J. Hum. Genet. 79 (4): 724–30. doi:10.1086/507792. PMC 1592572. PMID 16960809.