Allan–Herndon–Dudley syndrome
Allan–Herndon–Dudley syndrome is a rare X-linked inherited disorder of brain development that causes both moderate to severe intellectual disability and problems with speech and movement.[1]
| Allan–Herndon–Dudley syndrome | |
|---|---|
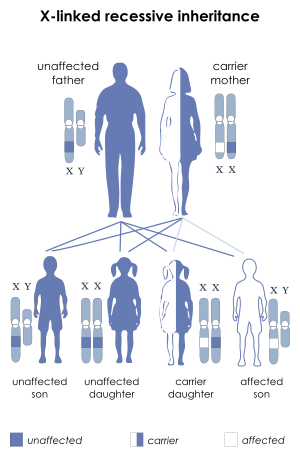 | |
| This condition is inherited in an X-linked recessive manner | |
| Specialty | Medical genetics, neurology |
Allan–Herndon–Dudley syndrome, which is named eponymously for William Allan, Florence C. Dudley, and C. Nash Herndon,[2][3] results from a mutation of the thyroid hormone transporter MCT8 (also referred to as SLC16A2). Consequently, thyroid hormones are unable to enter the nervous system, which depends on thyroid signaling for proper function and development.
Signs and symptoms
It is estimated that 80%-99% of people with Allan-Herndon-Dudley syndrome will have biparietal narrowing (narrowing of skull), ataxia, abnormalities of the neck, and both absent speech development and aphasia. Weak muscle tone (hypotonia) and underdevelopment of many muscles (muscle hypoplasia) are common in children with Allan-Herndon-Dudley syndrome. Development of joint deformities called contractures, which restrict the movement of certain joints, are common as people age. Mobility is further limited by abnormal muscle stiffness (spasticity), muscle weakness, and involuntary movements of the arms and legs. Many people with Allan–Herndon–Dudley syndrome are unable to walk independently and become wheelchair-bound by adulthood.[4]
Genetics
This condition is inherited in an X-linked recessive pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation must be present in both copies of the gene to cause the disorder. Males are affected by X-linked recessive disorders much more frequently than females. A striking characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In X-linked recessive inheritance, a female with one altered copy of the gene in each cell is called a carrier. She can pass on the mutated gene, but usually does not experience signs and symptoms of the disorder. Carriers of SLC16A2 mutations have normal intelligence and do not experience problems with movement. Some carriers have been diagnosed with thyroid disease, a condition which is relatively common in the general population. It is unclear whether thyroid disease is related to SLC16A2 mutations in these cases.
Pathogenesis
Mutations in the SLC16A2 gene cause Allan–Herndon–Dudley syndrome. The SLC16A2 gene, also known as MCT8, provides instructions for making a protein that plays a critical role in the development of the nervous system. This protein transports a particular hormone into nerve cells in the developing brain. This hormone, called triiodothyronine or T3, is produced by the thyroid. T3 appears to be critical for the normal formation and growth of nerve cells, as well as the development of junctions between nerve cells (synapses) where cell-to-cell communication occurs. T3 and other forms of thyroid hormone also help regulate the development of other organs and control the rate of chemical reactions in the body.
Gene mutations alter the structure and function of the SLC16A2 protein. As a result, this protein is unable to transport T3 into nerve cells effectively. A lack of this critical hormone in certain parts of the brain disrupts normal brain development, resulting in intellectual disability and problems with movement. Excess amounts of T3 circulate in the bloodstream. It is unclear if this is a consequence of compensatory hyperdeiodination or if it results from impaired uptake by certain cell types. Increased T3 levels in the blood may be toxic to some organs and contribute to the signs and symptoms of Allan–Herndon–Dudley syndrome.
Diagnosis
Treatment
In May 2013, the US FDA granted Orphan drug status to Diiodothyropropionic acid (DITPA) in the treatment of MCT8 deficiency. This was following the use of DITPA towards a child in Australia, under compassionate grounds.[5]
There is no established treatment for AHDS. Theoretical considerations suggested TRIAC (triiodothyroacetate or tiratricol, a natural non-classical thyroid hormone) to be beneficial. In 2014, a case was demonstrated in which therapy with TRIAC in early childhood led to significant improvement of cognition and mobility.[6] Currently, the effect of Triac is under investigation.[7]
References
- "Allan-Herndon-Dudley syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-04-17.
- synd/1438 at Who Named It?
- Allan, William; Herndon, C. N.; Dudley, Florence C. (1944). "Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly". American Journal of Mental Deficiency. 48: 325–34.
- "Allan-Herndon-Dudley syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-04-17.
- Verge, Charles F.; Konrad, Daniel; Cohen, Michal; Di Cosmo, Caterina; Dumitrescu, Alexandra M.; Marcinkowski, Teresa; Hameed, Shihab; Hamilton, Jill; Weiss, Roy E.; Refetoff, Samuel (2012). "Diiodothyropropionic Acid (DITPA) in the Treatment of MCT8 Deficiency". The Journal of Clinical Endocrinology & Metabolism. 97 (12): 4515–23. doi:10.1210/jc.2012-2556. PMC 3513545. PMID 22993035.
- Iglesias, Ainhoa; Palomares, María; Morte, Beatriz; Obregón, María Jesús; Bernal, Juan (September 10, 2014). TRIAC treatment of an infant with Allan-Herndon-Dudley Syndrome (AHDS): Effects on iodothyronines in serum and cerebrospinal fluid. 38th Annual Meeting of the European Thyroid Association. Santiago de Compostela. hdl:10261/125597.
- Clinical trial number NCT02060474 for "Triac Trial in MCT8 Patients" at ClinicalTrials.gov
External links
| Classification |
|---|
- GeneReviews/NCBI/NIH/UW entry on MCT8 (SLC16A2)-Specific Thyroid Hormone Cell Transporter Deficiency
- Allan–Herndon–Dudley syndrome at National Library of Medicine