Hawkinsinuria
Hawkinsinuria, is an autosomal dominant metabolic disorder affecting the metabolism of tyrosine.[1] Normally, the breakdown of the amino acid tyrosine involves the conversion of 4-hydroxyphenylpyruvate to homogentisate by 4-Hydroxyphenylpyruvate dioxygenase. Complete deficiency of this enzyme would lead to tyrosinemia III. In rare cases, however, the enzyme is still able to produce the reactive intermediate 1,2-epoxyphenyl acetic acid, but is unable to convert this intermediate to homogentisate. The intermediate then spontaneously reacts with glutathione to form 2-L-cystein-S-yl-1,4-dihydroxy-cyclohex-5-en-1-yl acetic acid (hawkinsin).
| Hawkinsinuria | |
|---|---|
| Other names | 4-Alpha-hydroxyphenylpyruvate hydroxylase deficiency |
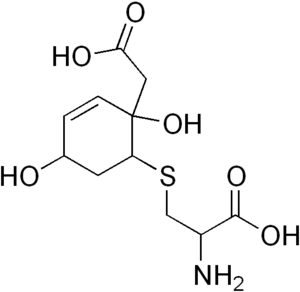 | |
| Hawkinsin | |
| Specialty | Endocrinology |
Patients present with metabolic acidosis during the first year of life (can manifest with metabolic acidosis and growth arrest around the time of weaning off breast milk) which should be treated by a phenylalanine- and tyrosine-restricted diet. The tolerance toward these amino acids normalizes as the patients get older. Then only a chlorine-like smell of the urine indicates the presence of the condition, patients have a normal life and do not require treatment or a special diet.
The production of hawkinsin is the result of a gain-of-function mutation, inheritance of hawkinsinuria is therefore autosomal dominant (presence of a single mutated copy of the gene causes the condition). Most other inborn errors of metabolism are caused by loss-of-function mutations, and hence have recessive inheritance (condition occurs only if both copies are mutated).
See also
- 4-Hydroxyphenylpyruvate dioxygenase
References
- Tomoeda K, Awata H, Matsuura T, Matsuda I, Ploechl E, Milovac T, Boneh A, Scott CR, Danks DM, Endo F (2000). "Mutations in the 4-hydroxyphenylpyruvic acid dioxygenase gene are responsible for tyrosinemia type III and hawkinsinuria". Mol Genet Metab. 71 (3): 506–510. doi:10.1006/mgme.2000.3085. PMID 11073718.