N-Acetylglutamate synthase deficiency
N-Acetylglutamate synthase deficiency is an autosomal recessive urea cycle disorder.
| N-Acetylglutamate synthase deficiency | |
|---|---|
| Other names | Synthetase deficiency |
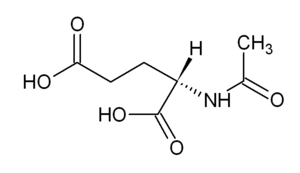 | |
| N-Acetylglutamic acid | |
| Specialty | Medical genetics |
Signs and symptoms
The symptoms are visible within the first week of life and if not detected and diagnosed correctly immediately consequences are fatal.
Genetics
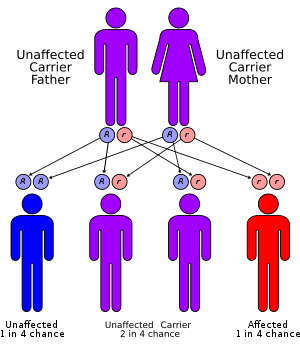
The chromosome found to be carrying the gene encoding for N-Acetyl Glutamate synthetase is chromosome 17q (q stands for longer arm of the chromosome) in humans and chromosome 11 in mice. In both organisms, the chromosome consists of seven exons and six introns and non-coding sequence.
The cause for this disorder is a single base deletion that led to frameshift mutation, and thus the error in gene's coding for this specific enzyme.
Mechanism
Carbamoyl phosphate synthase I is an enzyme found in mitochondrial matrix and it catalyzes the very first reaction of the Urea cycle, in which carbamoyl phosphate is produced.
Carbamoyl Phosphate Synthase 1, abbreviated as CPS1, is activated by its natural activator N-Acetyl glutamate, which in turn is synthesized from acetyl-CoA and glutamic acid in the reaction catalyzed by N-Acetyl glutamate synthase, commonly called NAGS. N-Acetyl Glutamate is required for the Urea cycle to take place.
Deficiency in N-Acetylglutamate synthase or a genetic mutation in the gene coding for the enzyme, will lead to urea cycle failure in which ammonia is not converted to urea, but rather accumulated in blood leading to the condition called Type I Hyperammonemia. This is a severe neonatal disorder with fatal consequences, if not detected immediately upon birth.
Diagnosis
Treatment
Although there is currently no cure, treatment includes injections of structurally similar compound, Carglumic acid, an analogue of N-Acetyl Glutamate. This analogue likewise activates CPS1. This treatment mitigates the intensity of the disorder.
If symptoms are detected early enough and the patient is injected with this compound, levels of severe mental retardation can be slightly lessened, but brain damage is irreversible.
Also: hemodialysis for emergent hyperammonemic crisis, Na benzoate, Na phenylacetate, Na phenylbutyrate, low-protein diet supplemented with essential amino acid mixture and arginine, citrulline, experimental attempts at gene therapy, liver transplantation (which is curative), and also N-carbamylglutamate supplementation.
See also
- Hyperammonemia
- Urea cycle
References
- Hall L, Metzenberg R, Cohen P (1958). "Isolation and characterization of a naturally occurring cofactor of carbamyl phosphate biosynthesis". J Biol Chem. 230 (2): 1013–21. PMID 13525417.
- Caldovic L, Morizono H, Panglao M, Cheng S, Packman S, Tuchman M (2003). "Null mutations in the N-acetylglutamate synthase gene associated with acute neonatal disease and hyperammonemia". Hum Genet. 112 (4): 364–8. doi:10.1007/s00439-003-0909-5. PMID 12594532.
External links
| Classification |
|
|---|---|
| External resources |