Syndrome de Prader-Willi
Le syndrome de Prader-Willi est un syndrome caractérisé à la naissance par une hypotonie sévère avec des difficultés alimentaires suivis par une hyperphagie responsable du développement d’une obésité morbide.
Tous les malades ont un comportement particulier avec des difficultés d’apprentissage.
L’hypogonadisme touche les deux sexes et la taille est réduite.
| Spécialité | Génétique médicale, pédiatrie et neurologie |
|---|
| CIM-10 | Q87.1 |
|---|---|
| CIM-9 | 759.81 |
| OMIM | 176270 |
| DiseasesDB | 10481 |
| MedlinePlus | 001605 |
| eMedicine | 947954 |
| eMedicine | ped/1880 |
| MeSH | D011218 |
| GeneReviews | |
| Médicament | Somatrem (en) |
| Patient UK | Prader-willi-syndrome-pro |
![]()
Le syndrome de Prader-Willi est classé parmi les maladies rares affectant un cas sur 25 000 naissances[1].
Étiologie
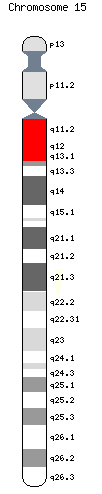
La cause de cette maladie est génétique. Alors que la plupart des gènes sont actifs ou inactifs de la même façon pour leurs deux copies, certains nécessitent que l'une des copies parentales soit réprimée, l'autre pouvant être exprimée. Cette répression génique s'appelle l'empreinte parentale. Cette empreinte est un cas particulier d'empreinte génétique, et la plus courante, pour laquelle le mode d'expression de chacune des copies n'est pas aléatoire mais dépend de leur origine, maternelle ou paternelle.
Dans le cas du syndrome de Prader-Willi, la région concernée (15q11-q13) se situe sur le bras long du chromosome 15. Sur le chromosome 15 d'origine paternelle, les gènes de la région 15q11-q13 sont fonctionnels, tandis qu'ils ne remplissent pas leur fonction sur le chromosome 15 d'origine maternelle (inactivation par méthylation)[2].
C’est l’absence de la région PWS/AS du chromosome 15 paternel qui est la cause de cette affection. Plusieurs mécanismes sont responsables de l’absence de région PWS/AS :
- dans 70 % des cas, délétion du locus q11.2-q13 du chromosome 15. Cette délétion ne concerne que le chromosome d’origine paternel ;
- dans 25 % des cas, disomie maternelle du chromosome 15. L’absence du chromosome 15 paternel empêche l’expression de la région PWS/AS ;
- dans 5 % des cas, il s’agit d’un défaut d’expression de la région PWS/AS bien que présente.
La délétion de la région PWS/AS du chromosome 15 maternel est responsable du syndrome d'Angelman.
Description
L’hypotonie à la naissance est un signe constant. Cette hypotonie s’accompagne d’une succion difficile, le cri est faible.
L’hypogonadisme est évident à la naissance surtout chez les garçons. Cet hypogonadisme d’origine centrale est responsable d’un retard de puberté et d’une infertilité.
Le retard de développement est aussi un signe constant, la marche est retardée.
La difficulté d’apprentissage devient évidente à l’école, le retard mental est léger à moyen mais la difficulté d’apprentissage n’est pas corrélée avec le retard mental.
Vers un an l’hyperphagie apparaît, aboutissant à une prise de poids très importante avec une tendance compulsive à satisfaire ses besoins alimentaires.
Le comportement psychologique caractéristique de ce syndrome apparaît tôt : troubles obsessionnels compulsifs, crises de colère, obstination, peur du changement.
La taille est petite avec des petites mains et des petits pieds.
Diagnostic
Le diagnostic clinique peut être fait selon les critères définis en 1999, chaque critère majeur vaut 1 point et chaque critère mineur vaut 0,5 point[3]. Avant 3 ans, cinq points sont nécessaires, dont quatre critères majeurs. Après 3 ans, huit points sont nécessaires, dont cinq critères majeurs.
Clinique
- Critères majeurs de diagnostic
- Hypotonie natale d’origine centrale avec succion faible s’améliorant avec l’âge
- Problème d’alimentation et/ou difficulté de croissance imposant des techniques alimentaires
- Gain très rapide de poids entre 12 mois et 6 ans entraînant une obésité morbide
- Hyperphagie
- Caractéristiques faciales
- Bouche éversée, diamètre frontal étroit
- Hypogonadisme
- Organes génitaux externes hypoplasiques
- Retard de la puberté
- Infertilité
- Retard d’apprentissage, difficulté d’apprentissage, retard mental moyen
- Critères mineurs de diagnostic
- Diminution des mouvements fœtaux et léthargie à la naissance
- Comportement typique
- Troubles obsessionnels compulsifs
- Crises de colère
- Obstination
- Tendance au vol et au mensonge
- Trouble du sommeil
- Petite taille pour la famille
- Hypopigmentation
- Petites mains et petits pieds
- Myopie
- Salive épaisse et visqueuse
- Difficultés d’articulation
- En raison de la nécessité d’un diagnostic rapide optimisant la prise en charge, la réalisation des tests génétiques pourra être réalisé sur les critères suivants en fonction de l’âge.
- Avant 2 ans
- Hypotonie natale d’origine centrale avec succion faible
- De 2 à 6 ans
- Hypotonie natale d’origine centrale avec succion faible
- Retard global de développement
- De 6 à 12 ans
- Histoire de succion faible
- Retard global de développement
- Gain très rapide de poids entraînant une obésité morbide
- À partir de 13 ans
- Difficulté d’apprentissage avec léger retard mental
- Hyperphagie avec obésité morbide
- Hypogonadisme hypogonadotrope avec comportement caractéristique.
- Avant 2 ans
Analyse chromosomique
La technique d'hybridation in situ par fluorescence ou l’étude en haute résolution de la bande 650 permet le diagnostic dans 70 % des cas.
Analyse génétique
Plus de 99 % des patients atteints de ce syndrome ont des anomalies isolées de la méthylation. Lorsqu’une anomalie de la méthylation est retrouvée, la stratégie de recherche de l’absence de la région PWS/AS dépend du mécanisme en cause.
Diagnostic différentiel
L’hypotonie à la naissance est aussi présente dans :
- Anomalie chromosomique (dup Xq27.2-ter, del 6q16.2, del 1p36, del 10q26)
- Syndrome de l'X fragile
- Syndrome de Rett
- Syndrome d'Angelman
L’association retard de développement, obésité et hypogonadisme se rencontre dans :
Traitement
À ce jour, il n'existe aucun traitement spécifique à ce syndrome. Seule la prévention des symptômes (hormones de croissance, régime équilibré, orthophonie, etc.) permet d'améliorer la qualité de vie des patients.
Notes et références
- Anonyme, Atténuer le syndrome de Prader-Willi, dans Le Monde du 5 septembre 2018, suppl. Science et Médecine
- « Le syndrome de Prader-Willi », sur Orpha.net (consulté le 4 décembre 2015).
- (en) Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY. & Greenberg F. « Prader-Willi Syndrome: Consensus diagnostic criteria » Pediatrics 1993;91:398-402.
Bibliographie
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 176270
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 182279
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 602117
- (en) Suzanne B Cassidy, Stuart Schwartz, Prader-Willi Syndrome in GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005
Liens externes
- Association Prader-Willi Belgique
- Association Prader-Willi France
- (fr/en) Fondation canadienne pour la recherche sur le syndrome de Prader-Willi
- Fiche résumé sur le Syndrome de Prader-Willi sur Orphanet

