Tetrasomy 18p
Tetrasomy 18p is a genetic condition that is caused by the presence of an isochromosome, composed of two copies of the short arm of chromosome 18. It is characterized by multiple medical and developmental concerns.[2]
| Tetrasomy 18p | |
|---|---|
| Other names | Isochromosome 18p[1] |
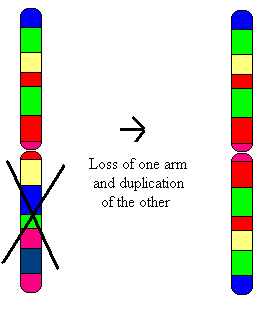 | |
| Condition is due to an isochromosome on 18 CHR | |
| Specialty | Medical genetics |
Signs and symptoms
Tetrasomy 18p causes a wide range of medical and developmental problems.[3]
Congenital anomalies
Cardiac anomalies are the most common congenital malformation in individuals with tetrasomy 18p. However, there is no pathognomatic heart defect associated with the condition. Patent ductus arteriosus is the most common defect. Septal defects (ventricular septal defects and atrial septal defects) are also common, as are patent foramina ovalia. Other cardiac anomalies include mitral valve regurgitation, mitral valve prolapse, bicuspid pulmonary valve, hypoplastic transverse aortic arch, tricuspid valve regurgitation, right ventricular hypertrophy, and pulmonic stenosis.
In males, cryptorchidism is common. Abnormal genitalia in females is not a common feature. Renal abnormalities have been reported in a minority of patients. Horseshoe kidney and bladder diverticuli have been reported. Other abdominal malformations, including pyloric stenosis and hernias, have also been reported, though they are present in only a minority of patients.
Orthopedic anomalies also occur relatively frequently, with hip dysplasia being the most common orthopedic issue. Clubfoot and rocker bottom feet have also been reported.
Myelomeningocele is another known feature associated with tetrasomy 18p.
Neonatal complications
Neonatal complications (apart from congenital anomalies) are common. In a paper published in 2010, 41 of 42 individuals had some type medical problem in the first days of life, the most common being feeding difficulties. Respiratory difficulty and jaundice are also relatively frequent.
ENT
Recurrent otitis media is common, and many patients required the placement of PE tubes. Small ear canals are also fairly common, but not as much as in 18q-.
Gastrointestinal
The most common gastrointestinal abnormality is chronic constipation, though gastrointestinal reflux was also common.
Endocrine
Growth hormone deficiency is present in about 20% of people with tetrasomy 18p.
Neurologic
Hypotonia and/or hypertonia are present in nearly all individuals with tetrasomy 18p. Approximately 25% have a seizure disorder.
Growth
Short stature is common, with ~50% being at or below the 25th centile and 20% being at or below the 3rd centile. Microcephaly is present in about 40% of patients.
Development
All reported cases of tetrasomy 18p have some degree of intellectual disability. Most individuals score in the moderate range of intellectual disability based on standardized testing.
Genetics
Tetrasomy 18p is caused by the presence of an additional isochromosome composed of two copies of the p arm of chromosome 18. It has been reported in both the non-mosaic as well as the mosaic state. (The phrase "mosaicism" in this context means that some cells carry the genetic change while others do not.) In the grand majority of cases, the isochromosome is de novo. Although there has been some speculation that tetrasomy 18p may occur with a higher frequency in children of older mothers,[4][5] there is not enough evidence to say that this is definitively the case.
Diagnosis
Suspicion of a chromosome abnormality is typically raised due to the presence of developmental delays or congenital malformations. Diagnosis of tetrasomy 18p is typically made via a routine chromosome analysis from a blood sample. The diagnosis can also be made prenatally by chorionic villus sampling or amniocentesis.
Severity of tetrasomy 18p is variable. Individuals with mosaicism are typically less severely affected than non-mosaic individuals.
Management
At present, treatment for tetrasomy 18p is symptomatic, meaning that the focus is on treating the signs and symptoms of the conditions as they arise. The Chromosome 18 Clinical Research Center has published a list of recommended screening and evaluations:[3]
- Genetics evaluation and counseling
- Parental chromosomes
- Periodic ophthalmology evaluations
- Periodic audiology evaluations
- ENT referral for management of chronic otitis media
- Cardiology evaluation
- Renal ultrasound
- Orthopedic evaluation for management of foot abnormalities
- Monitor for scoliosis and kyphosis
- Neurology evaluation for seizures, abnormal muscle tone
- Gastrointestinal/nutritional evaluation of failure to thrive, gastroesophageal reflux, constipation
- Endocrinology evaluation for short stature, to include evaluation for growth hormone deficiency
- Referral for developmental services and therapy
Research
Current research is focusing on clearly defining the phenotype associated with tetrasomy 18p and identifying which genes cause medical and developmental problems when present in four copies.
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Tetrasomy 18p". www.orpha.net. Retrieved 17 May 2019.
- Vazquez-Cantu, Diana (June 2018). "CLINICAL AND CYTOGENETIC CHARACTERIZATION OF A PATIENT WITH TETRASOMY 18P". Journal of Basic Applied Genetics. 29 (1): 17–23. Retrieved 12 March 2019.
- Sebold C, Roeder E, Zimmerman M, Soileau B, Heard P, Carter E, Schatz M, White WA, Perry B, Reinker K, O'Donnell L, Lancaster J, Li J, Hasi M, Hill A, Pankratz L, Hale DE, Cody JD (2010). Tetrasomy 18p: report of the molecular and clinical findings of 43 individuals. Am J Med Genet 152A(9): 2164-72.
- Bugge M, Blennow E, Friedrich U, Petersen MB, Pedeutour F, Tsezou A, Frum A, Hermann S, Lyngbye T, Sarri C, Avramopoulos D, Kitsiou S, Lambert JC, Guzda M, Tommerup N, Brondum-Nielsen K. 1996. Tetrasomy 18p de novo: Parental origin and different mechanisms of formation. Eur J Hum Genet 3:160–167.
- Kotzot D, Bundscherer G, Bernasconi F, Brecevic L, Lurie IW, Basaran S, Baccicchetti C, H€oller A, Castellan C, Braun-Quentin C, Pfeiffer RA, Schinzel A. 1996. Isochromosome 18p results from maternal meiosis II nondisjunction. Eur J Hum Genet 4:168–174.
External links
| Classification | |
|---|---|
| External resources |
|