Pyruvate dehydrogenase deficiency
Pyruvate dehydrogenase deficiency (also known as pyruvate dehydrogenase complex deficiency or PDCD) is one of the most common neurodegenerative disorders associated with abnormal mitochondrial metabolism. PDCD is an X-linked disease that shows heterogeneous characteristics in both clinical presentation and biochemical abnormality.[1] The pyruvate dehydrogenase complex (PDC) is a multi-enzyme complex that plays a vital role as a key regulatory step in the central pathways of energy metabolism in the mitochondria.
| Pyruvate dehydrogenase complex deficiency | |
|---|---|
| Specialty | Endocrinology |
Signs and symptoms
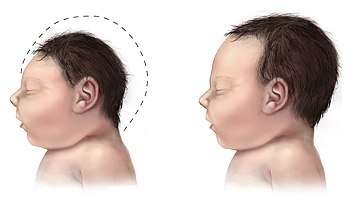
PDCD is generally presented in one of two forms. The metabolic form appears as lactic acidosis. The neurological form of PDCD contributes to hypotonia, poor feeding, lethargy and structural abnormalities in the brain.[2] Patients may develop seizures and/or neuropathological spasms. These presentations of the disease usually progress to mental retardation, microcephaly, blindness and spasticity.[3]
Females with residual pyruvate dehydrogenase activity will have no uncontrollable systemic lactic acidosis and few, if any, neurological symptoms. Conversely, females with little to no enzyme activity will have major structural brain abnormalities and atrophy. Males with mutations that abolish, or almost abolish, enzyme activity presumably die in utero because brain cells are not able to generate enough ATP to be functionally viable. It is expected that most cases will be of mild severity and have a clinical presentation involving lactic acidosis.[4]
Prenatal onset may present with non-specific signs such as low Apgar scores and small for gestational age. Metabolic disturbances may also be considered with poor feeding and lethargy out of proportion to a mild viral illness, and especially after bacterial infection has been ruled out.[3] PDH activity may be enhanced by exercise, phenylbutyrate and dichloroacetate.
The clinical presentation of congenital PDH deficiency is typically characterized by heterogenous neurological features that usually appear within the first year of life. In addition, patients usually show severe hyperventillation due to profound metabolic acidosis mostly related to lactic acidosis. Metabolic acidosis in these patients is usually refractory to correction with bicarbonate.[5]
Genetics
The most commonly seen form of PDCD is caused by mutations in the X-linked E1 alpha gene and is approximately equally prevalent in both males and females. However, a greater severity of symptoms tends to affect males more often than heterozygous females. This can be explained by x-inactivation, as females carry one normal and one mutant gene. Cells with a normal allele active can metabolize the lactic acid that is released by the PDH deficient cells. They cannot, however, supply ATP to these cells and, therefore, phenotype depends largely on the nature/severity of the mutation.[3][4]
Diagnosis

Pyruvate dehydrogenase deficiency can be diagnosed via the following methods:[6]
- Blood test (Lactate and pyruvate levels)
- Urine analysis
- Magnetic resonance spectroscopy
- MRI
Differential diagnosis
The differential diagnosis of pyruvate dehydrogenase deficiency can consist of either D-Lactic acidosis or abnormalities associated with gluconeogenesis.[6]
Treatment
Direct treatment that stimulates the pyruvate dehydrogenase complex (PDC), provides alternative fuels, and prevents acute worsening of the syndrome.[7] However, some correction of acidosis does not reverse all the symptoms. CNS damage is common and limits a full recovery. Ketogenic diets, with high fat and low carbohydrate intake have been used to control or minimize lactic acidosis and anecdotal evidence shows successful control of the disease, slowing progress and often showing rapid improvement. No study has yet been published demonstrating the effectiveness of the ketogenic diet for treatment of PDCD.
There is some evidence that dichloroacetate reduces the inhibitory phosphorylation of pyruvate dehydrogenase complex and thereby activates any residual functioning complex. Resolution of lactic acidosis is observed in patients with E1 alpha enzyme subunit mutations that reduce enzyme stability. However, treatment with dichloroacetate does not improve neurological damage.[3] Oral citrate is often used to treat acidosis.
See also
- Citric acid cycle
- Branched-chain alpha-keto acid dehydrogenase complex
- Anti-mitochondrial antibody
References
- Reference, Genetics Home. "pyruvate dehydrogenase deficiency". Genetics Home Reference. Retrieved 2016-11-08.
- Cassandra L. Kniffin (28 October 2014). "pyruvate dehydrogenase E1-alpha deficiency; PDHAD". Online Mendelian Inheritance in Man. Johns Hopkins University. Entry # 312170. Retrieved 2016-11-09.
- G K Brown; L J Otero; M LeGris; R M Brown (November 1994). "Pyruvate dehydrogenase deficiency". J Med Genet. 31 (11): 875–879. doi:10.1136/jmg.31.11.875. PMC 1016663. PMID 7853374.
- H H Dahl (March 1995). "Pyruvate dehydrogenase E1 alpha deficiency: males and females differ yet again". Am J Hum Genet. 56 (3): 553–557. PMC 1801181. PMID 7887408.
- Mitochondrion.
- "Pyruvate Dehydrogenase Complex Deficiency Workup: Laboratory Studies, Imaging Studies, Histologic Findings". emedicine.medscape.com. Retrieved 8 November 2016.
- Garry Brown, ed. (April 2012). "Pyruvate dehydrogenase deficiency". Orphanet. ORPHA765. Retrieved 8 November 2016.
External links
| Classification | |
|---|---|
| External resources |