Posterior polymorphous corneal dystrophy
Posterior Polymorphous Corneal Dystrophy (PPCD; sometimes also Schlichting dystrophy) is a type of corneal dystrophy, characterised by changes in Descemet's membrane and endothelial layer. Symptoms mainly consist of decreased vision due to corneal edema. In some cases they are present from birth, other patients are asymptomatic. Histopathological analysis shows that the cells of endothelium have some characteristics of epithelial cells and have become multilayered. The disease was first described in 1916 by Koeppe as keratitis bullosa interna.[1]
| Posterior polymorphous corneal dystrophy | |
|---|---|
| Other names | Ophthalmology |
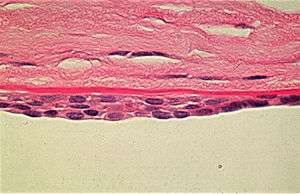 | |
| Appearance of the abnormal corneal endothelial cells that have become transformed into stratified squamous epithelium. Periodic acid Schiff (PAS) stain | |
Genetics
PPCD type 2 is linked to the mutations in COL8A2, and PPCD type 3 mutations in ZEB1 gene, but the underlying genetic disturbance in PPCD type 1 is unknown.
Pathophysiology
Vacuoles are demonstrated in the posterior parts of the cornea. The vesicles are located on the endothelial surface. The corneal endothelium is normally a single layer of cells that lose their mitotic potential after development is complete. In posterior polymorphous corneal dystrophy, the endothelium is often multilayered and has several other characteristics of an epithelium, including the presence of desmosomes, tonofilaments, and microvilli. These abnormal cells retain their ability to divide and extend onto the trabecular meshwork to cause glaucoma in up to 40% of cases.[2]
Diagnosis
See also
References
- Albrecht von Graefes (1916). "Klinische Beobachtungen mit der Nernstspaltlampe und dem Hornhautmikroskop". Arch. Klin. Exp. Ophthal. 91 (3): 363–379. doi:10.1007/BF01974655.
- "Posterior polymorphous corneal dystrophy". Online Mendelian Inheritance in Man(OMIM).