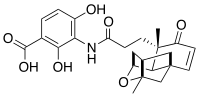
Platensimycin
Platensimycin, a metabolite of Streptomyces platensis, which is an excellent example of a unique structural class of natural antibiotics, has been demonstrated to be a breakthrough in recent antibiotic research due to its unique functional pattern and significant antibacterial activity.[1][2] This compound is a member of a class of antibiotics which act by blocking enzymes involved in the condensation steps in fatty acid biosynthesis,[3] which Gram-positive bacteria need to biosynthesise cell membranes (β-ketoacyl-(acyl-carrier-protein (ACP)) synthase I/II (FabF/B)). Other enzymes in this pathway have similarly been proven antibiotic targets for example FabI, the enoyl-ACP (acyl carrier protein) reductase, that is inhibited by isoniazid and related compounds and the antiseptic agent triclosan.[4]
 | |
 | |
| Names | |
|---|---|
| IUPAC name
3-[[3-[(1R,3R,4R,5aR,9R,9aS)-1,4,5,8,9,9a-Hexahydro-3,9-dimethyl-8-oxo-3H-1,4:3,5a-dimethano-2-benzoxepin-9-yl]-1-oxopropyl]amino]-2,4-dihydroxy-benzoic acid | |
| Identifiers | |
CAS Number |
|
3D model (JSmol) |
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank | |
PubChem CID |
|
InChI
| |
SMILES
| |
| Properties | |
Chemical formula |
C24H27NO7 |
| Molar mass | 441.480 g·mol−1 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
History
Platensimycin was first isolated from a strain of Streptomyces platensis by the Merck group [1] by using two-plate system where control organisms were compared to cells expressing fabF antisense RNA. This method uses the combination of target-based whole-cell and biochemical assays. The advantage of this method is that the concentrations of compounds in these extracts is sometimes too low to be identified in whole cell assays but are easily found in this two-plate assay system. They tried to find a drug targeting condensing enzymes and can be used clinically. Therefore, they systematically screened 250,000 natural product extracts (83,000 strains in three growth conditions) led to the identification of a potent and selective small molecule from a strain of Streptomyces platensis recovered from a soil sample collected in South Africa. This molecule, platensimycin (C24H27NO7, relative molecular mass 441.47), comprises two distinct structural elements connected by an amide bond. They still showed that Platensimycin has potent, broad-spectrum Gram-positive activity in vitro (Table below) and exhibits no cross-resistance to other key antibiotic-resistant bacteria including Methicillin-resistant Staphylococcus aureus(MRSA), vancomycin-intermediate S. aureus, vancomycin-resistant Enterococci, and linezolid-resistant and macrolide-resistant pathogens. [1] A first total synthesis of racemic platensimycin has been published.[7] Its structure consists of a 3-amino-2,4-dihydroxybenzoic acid polar part linked through an amide bond to a lipophilic tetracyclic ketolide.[5]
Clinical use
Platensimycin is an experimental new drug in preclinical trials in an effort to combat MRSA in a mouse model.[1] Platensimycin is a very effective antibiotic in vivo when continuously administered to cells, however this efficacy is reduced when administered by more conventional means.[6] Consequently, and in light of the elevated levels of the drug necessary for effectiveness, clinical trials have been delayed pending the development of variants of similar chemical form which have more favorable properties.[7] Since the importance of platensimycin, it has been studied to improve the production by gene regulation[8] and it is also emphasized recently to investigate the tolerance of modification without affecting activity by chemical modifications,[9] which can hopefully create the easier synthetic pathway[10][11] or increase the activity of platensimycin.[12]
Biosynthesis
The biosynthesis of plantensimycin has been studied by Singh Group[6] using isotope incorporation experiments to show that the benzoic ring is produced from pyruvate and acetate via the TCA cycle, while the C-17 tetracyclic enone acid core is produced from the non-mevalonate terpenoid pathway, the pathway is shown as below.[6]
Singh Group observed tetracyclic enone isotope labeling pattern is consistent with the biosynthesis of the tetracycle via the non-mevalonate terpenoid pathway proposed by Rohmer et al.[13] and Arigoni and co-workers.[14] This pathway involves condensation of a thiamine-activated acetyl group arising from the decarboxylation of pyruvate and glyceraldehyde-3-phosphate followed by a transposition step. Since both pyruvate and glyceraldehyde-3-phosphate (also glycerol) are part of the glycolytic pathway, varying levels of incorporations are expected. Thus, the terpenoid building blocks, dimethylallyl diphosphate and isopentenyl diphosphate, synthesized by the non-mevalonate pathway utilizing pyruvate and glyceraldehyde-3-phosphate, condense to form the diterpenoid precursor geranylgeranyl diphosphate that cyclizes to intermediate 3 which is related to (or derived from) ent-kaurene.[15] Oxidative cleavage of the double bond of intermediate would result in the loss of the terminal three carbons producing the C-17 tetracyclic enone acid unit. An N-acyltransferase reaction of tetracyclic enone and aminobenzoic acid would lead to platensimycin. Because of the dissimilarity between plantensimycin's mechanism of action and that of conventional antibiotics, it is believed that the capacity for the development of resistance by bacteria may be substantially decreased.[16]
Mechanism of action
Platensimycin has shown good activity against a panel of Gram positive organisms which included various resistant strains. Platensimycin works by inhibiting beta-ketoacyl synthases I/II (FabF/B) which are key enzymes in the production of fatty acids required for bacterial cell membranes. Here is the possible mechanism of action [2]: Firstly, the thiol group of FabF Cys163 is activated through the dipole moment of helix N-alpha-3 which lowers the pKa[17] of this functional group. The nucleophilicity of the cysteine is facilitated by an oxyanion hole formed with the backbone amides of Cys163 and Phe400. The crystal structure complex with platensimycin employed a C163Q mutant which gave a 50-fold increase in apparent binding [1]. The Gln163 residue lies adjacent to the carboxylate of platensimycin but makes no specific hydrogen bond. Given the close proximity of the carboxylate of platensimycin (presumed to be an anion) to the anionic thiol of Cys163 in the wild type enzyme may suggest the reason behind the increase in binding of the C163Q mutant. The second set of residues worth considering comprises His303 and His340 which play a role in the decarboxylation mechanism of the malonyl moiety. In particular, His303 activates a structured water to attack the carboxylate of the incoming malonyl-ACP.[18] The crystal structure of FabF also demonstrates that His340 forms a hydrogen bond between the amide nitrogen of Leu342 and the N-delta- atom of the imidazole ring meaning that the lone pair must reside on this atom [1,18]. In the platensimycin crystal structure the structured water adjacent to His303 is no longer present which may suggest an alternative electronic state for this residue. A strong possibility exists that His303 would present itself as a cation capable of forming an ionic interaction with the benzoic acid group of platensimycin [1].
References
- J. Wang, S.M. Soisson, K .Young, S.B. Singh etc,Nature 2006, 441, 358-61.
- D T. Manallack, I T. Crosby, Y. Khakham and B. Capuano, Current Medicinal Chemistry 2008, 15, 705-710.
- D. Häbich, F. Von Nussbaum, ChemMedChem 2006, 1, 951-954.
- H T. Wright, and K.A Reynolds, Curr Opin Microbiol 2007, 10, 447-53.
- K. C. Nicolaou, A. Li, D. J. Edmonds, Angew. Chem. 2006, 118, 7244-48.
- K B. Herath, A B. Attygalle, and S B. Singh, J. Am. Chem. Soc. 2007, 129, 15422-23.
- M J. Smanski, R M. Peterson, S R. Rajski, and B. Shen,Antimicrobial Agents and Chemotherapy. 2009
- Y. Chen, M.J. Smanski, B. Shen,Appl Microbiol Biotechnol 2010, 86, 19-25.
- J. Krauss, V. Knorr, V. Manhardt, S. Scheffels, F. Bracher, Arch Pharm (Weinheim) 2008, 341, 386-92.
- Y.Y. Yeung, E.J. Corey, Org Lett 2008, 10, 3877-8.
- X. Lu, Q. You, Curr Med Chem 2010, 17, 1139-55.
- D T. Manallack, I T. Crosby, Y. Khakham and B. Capuano, Current Medicinal Chemistry 2008, 15, 705-10.
- S. Smith, A. Witkowski, and A K. Joshi, Prog. Lipid Res. 2003, 42, 289-317.
- S W. White, J. Zheng, Y X M. Zhang, and C O. Rock, Annu. Rev. Biochem. 2005, 74, 791-831.
- W P. Revill, M J. Bibb, A K. Scheu, H J. Kieser, and D A. Hopwood, J. Bacteriol., 2001, 183, 3526-30.
- Potent antibiotic to target MRSA. BBC News. May 18, 2006. http://news.bbc.co.uk/1/hi/health/4992696.stm. Retrieved December 14,2009
- A C. Price, C O. Rock, S W. White, J. Bacteriol., 2003, 185, 4136-43.
- Y M. Zhang, J. Hurlbert, S W. White, C O. Rock, J. Biol. Chem. 2006, 281, 17390-99.