Neuroendocrine tumor
Neuroendocrine tumors (NETs) are neoplasms that arise from cells of the endocrine (hormonal) and nervous systems. Many are benign, while some are malignant. They most commonly occur in the intestine, where they are often called carcinoid tumors, but they are also found in the pancreas, lung and the rest of the body.
| Neuroendocrine tumor | |
|---|---|
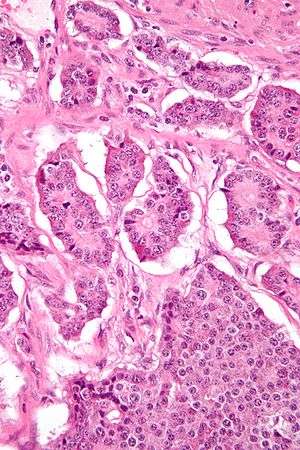 | |
| Micrograph of a neuroendocrine tumor. H&E stain | |
| Specialty | Endocrine oncology |
Although there are many kinds of NETs, they are treated as a group of tissue because the cells of these neoplasms share common features, such as looking similar, having special secretory granules, and often producing biogenic amines and polypeptide hormones.[1]
Classification
WHO
The World Health Organization (WHO) classification scheme places neuroendocrine tumors into three main categories, which emphasize the tumor grade rather than the anatomical origin:[2][3]
- well-differentiated neuroendocrine tumours, further subdivided into tumors with benign and those with uncertain behavior
- well-differentiated (low grade) neuroendocrine carcinomas with low-grade malignant behavior
- poorly differentiated (high grade) neuroendocrine carcinomas, which are the large cell neuroendocrine and small cell carcinomas.
Additionally, the WHO scheme recognizes mixed tumors with both neuroendocrine and epithelial carcinoma features, such as goblet cell cancer, a rare gastrointestinal tract tumor.[4]
Placing a given tumor into one of these categories depends on well-defined histological features: size, lymphovascular invasion, mitotic counts, Ki-67 labelling index, invasion of adjacent organs, presence of metastases and whether they produce hormones.[2][3]
Anatomic distribution
Traditionally, neuroendocrine tumors have been classified by their anatomic site of origin. NETs can arise in many different areas of the body, and are most often located in the intestine, pancreas or the lungs. The various kinds of cells that can give rise to NETs are present in endocrine glands and are also diffusely distributed throughout the body, most commonly Kulchitsky cells or similar enterochromaffin-like cells, that are relatively more common in the gastrointestinal and pulmonary systems.[5]
NETs include certain tumors of the gastrointestinal tract and of the pancreatic islet cells,[1] certain thymus and lung tumors, and medullary carcinoma of the parafollicular cells of the thyroid.[1] Tumors with similar cellular characteristics in the pituitary, parathyroid, and adrenomedullary glands are sometimes included[6] or excluded.[1]
Within the broad category of neuroendocrine tumors there are many different tumor types:[7] this outline is presented to facilitate retrieving information. Neuroendocrine tumors are uncommon in many of these areas, and frequently represent only a very small proportion of the tumors or cancers at these locations.
- Pituitary gland: Neuroendocrine tumor of the anterior pituitary
- Thyroid gland: Neuroendocrine thyroid tumors, particularly medullary carcinoma
- Parathyroid tumors
- Thymus and mediastinal carcinoid tumors[8][9]
- Pulmonary neuroendocrine tumors[10][11]
- bronchus[9]
- pulmonary carcinoid tumors: typical carcinoid (TC; low-grade); atypical carcinoid (AC; intermediate-grade)
- small-cell lung cancer (SCLC)
- large cell neuroendocrine carcinoma of the lung (LCNEC)[12]
- Extrapulmonary small cell carcinomas (ESCC or EPSCC)
- Gastroenteropancreatic neuroendocrine tumors (GEP-NET)[13][14]
- Foregut GEP-NET (foregut tumors can conceptually encompasses not only NETs of the stomach and proximal duodenum, but also the pancreas, and even thymus, lung and bronchus)
- Pancreatic endocrine tumors (if considered separately from foregut GEP-NET)[15]
- Midgut GEP-NET (from distal half of 2nd part of the duodenum to the proximal two-thirds of the transverse colon)
- appendix,[16] including well differentiated NETs (benign); well differentiated NETs (uncertain malignant potential); well differentiated neuroendocrine carcinoma (with low malignant potential); mixed exocrine-neuroendocrine carcinoma (goblet cell carcinoma, also called adenocarcinoid and mucous adenocarcinoid)
- Hindgut GEP-NET[17][18]
- Foregut GEP-NET (foregut tumors can conceptually encompasses not only NETs of the stomach and proximal duodenum, but also the pancreas, and even thymus, lung and bronchus)
- Liver[19][20][21] and gallbladder[22]
- Adrenal tumors, particularly adrenomedullary tumors
- Pheochromocytoma
- Peripheral nervous system tumors, such as:
- Breast[23]
- Genitourinary tract
- Merkel cell carcinoma of skin (trabecular cancer)
- Several inherited conditions:[29]
Grading
Neuroendocrine lesions are graded histologically according to markers of cellular proliferation, rather than cellular polymorphism. The following grading scheme is currently recommended for all gastroenteropancreatic neuroendocrine neoplasms by the World Health Organization:[35]
| G | Mitotic count (per 10 HPF) | Ki-67 index (%) |
|---|---|---|
| GX | Grade cannot be assessed | |
| G1 | < 2 | < 3% |
| G2 | 2 to 20 | 3% - 20% |
| G3 | > 20 | > 20% |
If mitotic count and Ki-67 are discordant, the figure which gives the highest grade is used.
G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) - formerly called carcinoid tumours. G3 neoplasms are called neuroendocrine carcinomas (NECs).
It has been proposed that the current G3 category be further separated into histologically well-differentiated and poorly-differentiated neoplasms to better reflect prognosis.[36]
Staging
Currently there is no one staging system for all neuroendocrine neoplasms. Well differentiated lesions generally have their own staging system based on anatomical location, whereas poorly differentiated and mixed lesions are staged as carcinomas of that location. For example, gastric NEC and mixed adenoneuroendocrine cancers are staged as a primary carcinoma of the stomach.[37]
TNM staging of gastroenteropancreatic Grade 1 and Grade 2 neuroendocrine tumors is as follows:
| Primary Tumor (T) | |
|---|---|
| T Category | Tumor Criteria |
| TX | Primary tumour cannot be assessed |
| T0 | No evidence of primary tumour |
| T1 | Invades the lamina propria or submucosa, and less than or equal to 1 cm in size |
| T2 | Invades the muscularis propria, or greater than 1 cm in size |
| T3 | Invades through the muscularis propria into subserosal tissue without penetration of overlying serosa |
| T4 | Invades visceral peritoneum (serosal) or other organs or adjacent structures |
| Regional Lymph Node (N) | |
| N Category | N Criteria |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
| Distant Metastasis (M) | |
| M Category | M Criteria |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Metastasis confined to liver |
| M1b | Metastasis in at least one extra-hepatic site |
| M1c | Both hepatic and extra-hepatic metastases |
| AJCC Prognostic Stage Groups | |
| Stage | Criteria |
| I | T1, N0, M0 |
| II | T2 or T3, N0, M0 |
| III | Any T, N1, M0; T4, N0, M0 |
| IV | Any T, any N, M1 |
| Primary Tumor (T) | |
|---|---|
| T Category | Tumor Criteria |
| TX | Primary tumour cannot be assessed |
| T1 | Invades the mucosa or submucosa only, and less than or equal to 1 cm in size (duodenal tumors) Confined within the sphincter of Oddi, and less than or equal to 1 cm in size (ampullary tumors) |
| T2 | Invades the muscularis propria, or is > 1 cm (duodenal) Invades through sphincter into duodenal submucosa or muscularis propria, or is > 1 cm (ampullary) |
| T3 | Invades the pancreas or peripancreatic adipose tissue |
| T4 | Invades visceral peritoneum (serosal) or other organs |
| Regional Lymph Node (N) | |
| N Category | N Criteria |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
| Distant Metastasis (M) | |
| M Category | M Criteria |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Metastasis confined to liver |
| M1b | Metastasis in at least one extra-hepatic site |
| M1c | Both hepatic and extra-hepatic metastases |
| AJCC Prognostic Stage Groups | |
| Stage | Criteria |
| I | T1, N0, M0 |
| II | T2 or T3, N0, M0 |
| III | T4, N0, M0; Any T, N1, M0 |
| IV | Any T, any N, M1 |
| Primary Tumor (T) | |
|---|---|
| T Category | Tumor Criteria |
| TX | Primary tumour cannot be assessed |
| T0 | No evidence of primary tumour |
| T1 | Invades the lamina propria or submucosa, and less than or equal to 1 cm in size |
| T2 | Invades the muscularis propria, or greater than 1 cm in size |
| T3 | Invades through the muscularis propria into subserosal tissue without penetration of overlying serosa |
| T4 | Invades visceral peritoneum (serosal) or other organs or adjacent structures |
| Regional Lymph Node (N) | |
| N Category | N Criteria |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis less than 12 nodes |
| N2 | Large mesenteric masses (> 2 cm) and / or extensive nodal deposits (12 or greater), especially those that encase the superior mesenteric vessels |
| Distant Metastasis (M) | |
| M Category | M Criteria |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Metastasis confined to liver |
| M1b | Metastasis in at least one extra-hepatic site |
| M1c | Both hepatic and extra-hepatic metastases |
| AJCC Prognostic Stage Groups | |
| Stage | Criteria |
| I | T1, N0, M0 |
| II | T2 or T3, N0, M0 |
| III | Any T, N1 or N2, M0; T4, N0, M0; |
| IV | Any T, any N, M1 |
| Primary Tumor (T) | |
|---|---|
| T Category | Tumor Criteria |
| TX | Primary tumour cannot be assessed |
| T0 | No evidence of primary tumour |
| T1 | 2 cm or less in greatest dimension |
| T2 | Tumor more than 2 cm but less than or equal to 4 cm |
| T3 | Tumor more than 4 cm or with subserosal invasion or involvement of the mesoappendix |
| T4 | Perforates the peritoneum or directly invades other organs or structures (excluding direct mural extension to adjacent subserosa of adjacent bowel) |
| Regional Lymph Node (N) | |
| N Category | N Criteria |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
| Distant Metastasis (M) | |
| M Category | M Criteria |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Metastasis confined to liver |
| M1b | Metastasis in at least one extra-hepatic site |
| M1c | Both hepatic and extra-hepatic metastases |
| AJCC Prognostic Stage Groups | |
| Stage | Criteria |
| I | T1, N0, M0 |
| II | T2 or T3, N0, M0 |
| III | Any T, N1, M0; T4, N1, M0 |
| IV | Any T, any N, M1 |
| Primary Tumor (T) | |
|---|---|
| T Category | Tumor Criteria |
| TX | Primary tumour cannot be assessed |
| T0 | No evidence of primary tumour |
| T1 | Invades the lamina propria or submucosa, and less than or equal to 2 cm |
| T1a | Less than 1 cm in greatest dimension |
| T1b | 1–2 cm in greatest dimension |
| T2 | Invades the muscularis propria, or greater than 2 cm in size with invasion of the lamina propria or submucosa |
| T3 | Invades through the muscularis propria into subserosal tissue without penetration of overlying serosa |
| T4 | Invades visceral peritoneum (serosal) or other organs or adjacent structures |
| Regional Lymph Node (N) | |
| N Category | N Criteria |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
| Distant Metastasis (M) | |
| M Category | M Criteria |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Metastasis confined to liver |
| M1b | Metastasis in at least one extra-hepatic site |
| M1c | Both hepatic and extra-hepatic metastases |
| AJCC Prognostic Stage Groups | |
| Stage | Criteria |
| I | T1, N0, M0 |
| IIA | T2, N0, M0 |
| IIB | T3, N0, M0 |
| IIIA | T4, N0, M0 |
| IIIB | Any T, N1, M0 |
| IV | Any T, any N, M1 |
| Primary Tumor (T) | |
|---|---|
| T Category | Tumor Criteria |
| TX | Primary tumour cannot be assessed |
| T1 | Limited to the pancreas, less than or equal to 2 cm in size |
| T2 | Limited to the pancreas, 2 – 4 cm in size |
| T3 | Limited to the pancreas, > 4 cm; or invading the duodenum or bile duct |
| T4 | Invading adjacent organs or the wall of large vessels |
| Regional Lymph Node (N) | |
| N Category | N Criteria |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node involvement |
| N1 | Regional lymph node involvement |
| Distant Metastasis (M) | |
| M Category | M Criteria |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Metastasis confined to liver |
| M1b | Metastasis in at least one extra-hepatic site |
| M1c | Both hepatic and extra-hepatic metastases |
| AJCC Prognostic Stage Groups | |
| Stage | Criteria |
| I | T1, N0, M0 |
| II | T2 or T3, N0, M0 |
| III | Any T, N1, M0; T4, N0, M0 |
| IV | Any T, any N, M1 |
Signs and symptoms
Gastroenteropancreatic neuroendocrine tumors (GEP-NET)
Conceptually, there are two main types of NET within this category: those which arise from the gastrointestinal (GI) system and those that arise from the pancreas. In usage, the term "carcinoid" has often been applied to both, although sometimes it is restrictively applied to NETs of GI origin (as herein), or alternatively to those tumors which secrete functional hormones or polypeptides associated with clinical symptoms, as discussed.
Carcinoid tumors
Carcinoids most commonly affect the small bowel, particularly the ileum, and are the most common malignancy of the appendix. Many carcinoids are asymptomatic and are discovered only upon surgery for unrelated causes. These coincidental carcinoids are common; one study found that one person in ten has them.[44] Many tumors do not cause symptoms even when they have metastasized.[45] Other tumors even if very small can produce adverse effects by secreting hormones.[46]
Ten per cent (10%)[47] or less of carcinoids, primarily some midgut carcinoids, secrete excessive levels of a range of hormones, most notably serotonin (5-HT) or substance P,[48] causing a constellation of symptoms called carcinoid syndrome:
- flushing
- diarrhea
- asthma or wheezing
- congestive heart failure (CHF)
- abdominal cramping
- peripheral edema
- heart palpitations
A carcinoid crisis with profound flushing, bronchospasm, tachycardia, and widely and rapidly fluctuating blood pressure[1] can occur if large amounts of hormone are acutely secreted,[48] which is occasionally triggered by factors such as diet,[48] alcohol,[48] surgery[1][48] chemotherapy,[48] embolization therapy or radiofrequency ablation.[1]
Chronic exposure to high levels of serotonin causes thickening of the heart valves, particularly the tricuspid and the pulmonic valves, and over a long period can lead to congestive heart failure.[48] However, valve replacement is rarely needed.[49] The excessive outflow of serotonin can cause a depletion of tryptophan leading to niacin deficiency, and thus pellagra,[1] which is associated with dermatitis, dementia, and diarrhea. Many other hormones can be secreted by some of these tumors, most commonly growth hormone that can cause acromegaly, or cortisol, that can cause Cushing's syndrome.
Occasionally, haemorrhage or the effects of tumor bulk are the presenting symptoms. Bowel obstruction can occur, sometimes due to fibrosing effects of NET secretory products[46] with an intense desmoplastic reaction at the tumor site, or of the mesentery.
Pancreatic neuroendocrine tumors
Pancreatic neuroendocrine tumors (PanNETs) are often referred to as "islet cell tumors",[50][51] or "pancreatic endocrine tumors"[2]
The PanNET denomination is in line with current WHO guidelines. Historically, PanNETs have also been referred to by a variety of terms, and are still often called "islet cell tumors" or "pancreatic endocrine tumors".[52] originate within the pancreas. PanNETs are quite distinct from the usual form of pancreatic cancer, adenocarcinoma, which arises in the exocrine pancreas. About 95 percent of pancreatic tumors are adenocarcinoma; only 1 or 2% of clinically significant pancreas neoplasms are GEP-NETs.
Well or intermediately differentiated PanNETs are sometimes called islet cell tumors; neuroendocrine cancer (NEC) (synonymous with islet cell carcinoma) is more aggressive. Up to 60% of PanNETs are nonsecretory or nonfunctional, which either don’t secrete, or the quantity or type of products such as pancreatic polypeptide (PPoma), chromogranin A, and neurotensin do not cause a clinical syndrome, although blood levels may be elevated.[29] Functional tumors are often classified by the hormone most strongly secreted by the pancreatic neuroendocrine tumor, as discussed in that main article.
Other
In addition to the two main categories of GEP-NET, there are rarer forms of neuroendocrine tumors that arise anywhere in the body, including within the lung, thymus and parathyroid. Bronchial carcinoid can cause airway obstruction, pneumonia, pleurisy, difficulty with breathing, cough, and hemoptysis, or may be associated with weakness, nausea, weight loss, night sweats, neuralgia, and Cushing's syndrome. Some are asymptomatic.
Animal neuroendocrine tumors include neuroendocrine cancer of the liver in dogs, and devil facial tumor disease in Tasmanian devils.[53][54][55]
Familial syndromes
Most pancreatic NETs are sporadic.[50] However, neuroendocrine tumors can be seen in several inherited familial syndromes, including:[29]
- multiple endocrine neoplasia type 1 (MEN1)
- multiple endocrine neoplasia type 2 (MEN2)
- von Hippel-Lindau (VHL) disease[29]
- neurofibromatosis type 1[30]
- tuberous sclerosis[31][32]
- Carney complex[33][34]
Given these associations, recommendations in NET include family history evaluation, evaluation for second tumors, and in selected circumstances testing for germline mutations such as for MEN1.[1]
Pathophysiology
NETs are believed to arise from various neuroendocrine cells whose normal function is to serve at the neuroendocrine interface. Neuroendocrine cells are present not only in endocrine glands throughout the body that produce hormones, but are found in all body tissues.[56]
Diagnosis
Markers
Symptoms from secreted hormones may prompt measurement of the corresponding hormones in the blood or their associated urinary products, for initial diagnosis or to assess the interval change in the tumor. Secretory activity of the tumor cells is sometimes dissimilar to the tissue immunoreactivity to particular hormones.[57]
Given the diverse secretory activity of NETs there are many other potential markers, but a limited panel is usually sufficient for clinical purposes.[1] Aside from the hormones of secretory tumors, the most important markers are:
- chromogranin A (CgA), present in 99% of metastatic carcinoid tumors[58]
- urine 5-hydroxyindoleacetic acid (5-HIAA)
- neuron-specific enolase (NSE, gamma-gamma dimer)
- synaptophysin (P38)
Newer markers include N-terminally truncated variant of Hsp70 is present in NETs but absent in normal pancreatic islets.[59] High levels of CDX2, a homeobox gene product essential for intestinal development and differentiation, are seen in intestinal NETs. Neuroendocrine secretory protein-55, a member of the chromogranin family, is seen in pancreatic endocrine tumors but not intestinal NETs.[59]
Imaging
CT-scans, MRIs, sonography (ultrasound), and endoscopy (including endoscopic ultrasound) are common diagnostic tools. CT-scans using contrast medium can detect 95 percent of tumors over 3 cm in size, but generally not tumors under 1 cm.[3]
Advances in nuclear medicine imaging, also known as molecular imaging, has improved diagnostic and treatment paradigms in patients with neuroendocrine tumors. This is because of its ability to not only identify sites of disease but also characterize them. Neuronedocrine tumours express somatostatin receptors providing a unique target for imaging. Octreotide is a synthetic modifications of somatostatin with a longer half-life. OctreoScan, also called somatostatin receptor scintigraphy (SRS or SSRS), utilizes intravenously administered octreotide that is chemically bound to a radioactive substance, often indium-111, to detect larger lesions with tumor cells that are avid for octreotide.
Somatostatin receptor imaging can now be performed with positron emission tomography (PET) which offers higher resolution, three-dimensional and more rapid imaging. Gallium-68 receptor PET-CT is much more accurate than an OctreoScan.[60]
Imaging with fluorine-18 fluorodeoxyglucose (FDG) PET may be valuable to image some neuroendocrine tumors.[61] This scan is performed by injected radioactive sugar intravenously. Tumors that grow more quickly use more sugar. Using this scan, the aggressiveness of the tumor can be assessed.
The combination of somatostatin receptor and FDG PET imaging is able to quantify somatostatin receptor cell surface (SSTR) expression and glycolytic metabolism, respectively.[61] The ability to perform this as a whole body study is highlighting the limitations of relying on histopathology obtained from a single site. This is enabling better selection of the most appropriate therapy for an individual patient.[62]
Histopathology
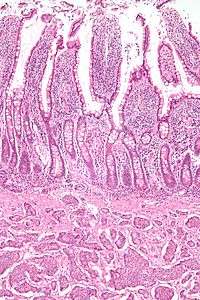
Features in common
Neuroendocrine tumors, despite differing embryological origin, have common phenotypic characteristics. NETs show tissue immunoreactivity for markers of neuroendocrine differentiation (pan-neuroendocrine tissue markers) and may secrete various peptides and hormones. There is a lengthy list of potential markers in neuroendocrine tumors; several reviews provide assistance in understanding these markers.[63][57] Widely used neuroendocrine tissue markers are various chromogranins, synaptophysin and PGP9.5. Neuron-specific enolase (NSE) is less specific.[1][5] The nuclear neuroendocrine marker insulinoma-associated protein-1 (INSM1) has proven to be sensitive as well as highly specific for neuroendocrine differentiation.[64]
NETs are often small, yellow or tan masses, often located in the submucosa or more deeply intramurally, and they can be very firm due to an accompanying intense desmoplastic reaction. The overlying mucosa may be either intact or ulcerated. Some GEP-NETs invade deeply to involve the mesentery. Histologically, NETs are an example of "small blue cell tumors," showing uniform cells which have a round to oval stippled nucleus and scant, pink granular cytoplasm. The cells may align variously in islands, glands or sheets. High power examination shows bland cytopathology. Electron microscopy can identify secretory granules. There is usually minimal pleomorphism but less commonly there can be anaplasia, mitotic activity, and necrosis.
Some neuroendocrine tumor cells possess especially strong hormone receptors, such as somatostatin receptors and uptake hormones strongly. This avidity can assist in diagnosis and may make some tumors vulnerable to hormone targeted therapies.
Argentaffin and hormone secretion
NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut; individual tumors within these sites can differ from these group benchmarks:
- Foregut NETs are argentaffin negative. Despite low serotonin content, they often secrete 5-hydroxytryptophan (5-HTP), histamine, and several polypeptide hormones. There may be associated atypical carcinoid syndrome, acromegaly, Cushing disease, other endocrine disorders, telangiectasia, or hypertrophy of the skin in the face and upper neck.[65] These tumors can metastasize to bone.
- Midgut NETs are argentaffin positive, can produce high levels of serotonin 5-hydroxytryptamine (5-HT), kinins, prostaglandins, substance P (SP), and other vasoactive peptides, and sometimes produce corticotropic hormone (previously adrenocorticotropic hormone [ACTH]). Bone metastasis is uncommon.
- Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. Bone metastases are not uncommon.
Treatment
Several issues help define appropriate treatment of a neuroendocrine tumor, including its location, invasiveness, hormone secretion, and metastasis. Treatments may be aimed at curing the disease or at relieving symptoms (palliation). Observation may be feasible for non-functioning low grade neuroendocrine tumors. If the tumor is locally advanced or has metastasized, but is nonetheless slowly growing, treatment that relieves symptoms may often be preferred over immediate challenging surgeries.
Intermediate and high grade tumors (noncarcinoids) are usually best treated by various early interventions (active therapy) rather than observation (wait-and-see approach).[66]
Treatments have improved over the past several decades, and outcomes are improving.[46] In malignant carcinoid tumors with carcinoid syndrome, the median survival has improved from two years to more than eight years.[67]
Detailed guidelines for managing neuroendocrine tumors are available from ESMO,[68] NCCN[69] and a UK panel.[1] The NCI has guidelines for several categories of NET: islet cell tumors of the pancreas,[70] gastrointestinal carcinoids,[71] Merkel cell tumors[72] and pheochromocytoma/paraganglioma.[73]
Surgery
Even if the tumor has advanced and metastasized, making curative surgery infeasible, surgery often has a role in neuroendocrine cancers for palliation of symptoms and possibly increased lifespan.[66]
Cholecystectomy is recommended if there is a consideration of long-term treatment with somatostatin analogs.[74]:46
Symptomatic relief
In secretory tumors, somatostatin analogs given subcutaneously or intramuscularly alleviate symptoms by blocking hormone release. A consensus review has reported on the use of somatostatin analogs for GEP-NETs.[75]
These medications may also anatomically stabilize or shrink tumors, as suggested by the PROMID study (Placebo-controlled prospective randomized study on the antiproliferative efficacy of Octreotide LAR in patients with metastatic neuroendocrine MIDgut tumors): at least in this subset of NETs, average tumor stabilization was 14.3 months compared to 6 months for placebo.[76]
The CLARINET study (a randomized, double-blind, placebo-controlled study on the antiproliferative effects of lanreotide in patients with enteropancreatic neuroendocrine tumors) further demonstrated the antiproliferative potential of lanreotide, a somatostatin analog and recently approved FDA treatment for GEP-NETS. In this study, lanreotide showed a statistically significant improvement in progression-free survival, meeting its primary endpoint. The disease in sixty five percent of patients treated with lanreotide in the study had not progressed or caused death at 96 weeks, the same was true of 33% of patients on placebo. This represented a 53% reduction in risk of disease progression or death with lanreotide based on a hazard ratio of .47.[77]
Lanreotide is the first and only FDA approved antitumor therapy demonstrating a statistically significant progression-free survival benefit in a combined population of patients with GEP-NETS.
Other medications that block particular secretory effects can sometimes relieve symptoms.[49]
Chemotherapy
Interferon is sometimes used to treat GEP-NETs.[78] Its effectiveness is somewhat uncertain, but low doses can be titrated within each person, often considering the effect on the blood leukocyte count;[78] Interferon is often used in combination with other agents, especially somatostatin analogs such as octreotide.
Gastrointestinal neuroendocrine tumors
Most gastrointestinal carcinoid tumors tend not to respond to chemotherapy agents,[49] showing 10 to 20% response rates that are typically less than 6 months. Combining chemotherapy medications has not usually been of significant improvement[49] showing 25 to 35% response rates that are typically less than 9 months.
The exceptions are poorly differentiated (high-grade or anaplastic) metastatic disease, where cisplatin with etoposide may be used[49] and Somatostatin Receptor Scintigraphy (SSRS) negative tumors which had a response rate in excess of 70% compared to 10% in strongly positive SRSS carcinoid tumors.[1]
PanNETs
Targeted therapy with everolimus (Afinitor) and sunitinib (Sutent) is approved by the FDA in unresectable, locally advanced or metastatic PanNETs. Some PanNETs are more responsive to chemotherapy than gastroenteric carcinoid tumors. Several agents have shown activity[49] and combining several medicines, particularly doxorubicin with streptozocin and fluorouracil (5-FU or f5U), is often more effective. Although marginally effective in well-differentiated PETs, cisplatin with etoposide is active in poorly differentiated neuroendocrine cancers (PDNECs).[49]
Radionuclide therapy
Peptide receptor radionuclide therapy (PRRT) is a type of radioisotope therapy (RIT)[6] in which a peptide or hormone conjugated to a radionuclide or radioligand is given intravenously, the peptide or neuroamine hormone previously having shown good uptake of a tracer dose, using Somatostatin receptor imaging as detailed above. This type of radiotherapy is a systemic therapy and will impact somatostatin positive disease.[79] The peptide receptor may be bound to lutetium-177, yttrium-90, indium-111 and other isotopes including alpha emitters.[80] This is a highly targeted and effective therapy with minimal side effects in tumors with high levels of somatostatin cell surface expression, because the radiation is absorbed at the sites of the tumor, or excreted in the urine. The radioactively labelled hormones enter the tumor cells which, together with nearby cells, are damaged by the attached radiation. Not all cells are immediately killed; cell death can go on for up to two years.
PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs[81][82] provided they demonstrate high uptake on SSTR imaging to suggest benefit.
Hepatic artery
Metastases to the liver can be treated by several types of hepatic artery treatments based on the observation that tumor cells get nearly all their nutrients from the hepatic artery, while the normal cells of the liver get about 70–80 percent of their nutrients and 50% their oxygen supply from the portal vein, and thus can survive with the hepatic artery effectively blocked.[46][83]
- Hepatic artery embolization (HAE) occludes the blood flow to the tumors, achieving significant tumor shrinkage in over 80%.[48] In hepatic artery chemotherapy, the chemotherapy agents are given into the hepatic artery, often by steady infusion over hours or even days. Compared with systemic chemotherapy, a higher proportion of the chemotherapy agents are (in theory) delivered to the lesions in the liver.[83]
- Hepatic artery chemoembolization (HACE), sometimes called transarterial chemoembolization (TACE), combines hepatic artery embolization with hepatic artery chemoinfusion: embospheres bound with chemotherapy agents, injected into the hepatic artery, lodge in downstream capillaries. The spheres not only block blood flow to the lesions, but by halting the chemotherapy agents in the neighborhood of the lesions, they provide a much better targeting leverage than chemoinfusion provides.
- Selective internal radiation therapy (SIRT)[84] for neuroendocrine metastases to the liver[85] delivers radioactive microsphere therapy (RMT) by injection into the hepatic artery, lodging (as with HAE and HACE) in downstream capillaries. In contrast to hormone-delivered radiotherapy, the lesions need not overexpress peptide receptors. The mechanical targeting delivers the radiation from the yttrium-labeled microspheres selectively to the tumors without unduly affecting the normal liver.[86] This type of treatment is FDA approved for liver metastases secondary to colorectal carcinoma and is under investigation for treatment of other liver malignancies, including neuroendocrine malignancies.[84]
Other therapies
Radiofrequency ablation (RFA) is used when a patient has relatively few metastases. In RFA, a needle is inserted into the center of the lesion and current is applied to generate heat; the tumor cells are killed by cooking.
Cryoablation is similar to RFA; an endothermic substance is injected into the tumors to kill by freezing. Cryoablation has been less successful for GEP-NETs than RFA.
AdVince, a type of gene therapy using a genetically modified oncolytic adenovirus[87] and supported by the crowdfunding campaign iCancer[88] was used in a Phase 1 trial against NET in 2016.[89]
Incidence
Although estimates vary, the annual incidence of clinically significant neuroendocrine tumors is approximately 2.5–5 per 100,000;[90] two thirds are carcinoid tumors and one third are other NETs.
The prevalence has been estimated as 35 per 100,000,[90] and may be considerably higher if clinically silent tumors are included. An autopsy study of the pancreas in people who died from unrelated causes discovered a remarkably high incidence of tiny asymptomatic NETs. Routine microscopic study of three random sections of the pancreas found NETs in 1.6%, and multiple sections identified NETs in 10%.[91] As diagnostic imaging increases in sensitivity, such as endoscopic ultrasonography, very small, clinically insignificant NETs may be coincidentally discovered; being unrelated to symptoms, such neoplasms may not require surgical excision.
History
Small intestinal neuroendocrine tumors were first distinguished from other tumors in 1907.[92][45] They were named carcinoid tumors because their slow growth was considered to be "cancer-like" rather than truly cancerous.[45]
However, in 1938 it was recognized that some of these small bowel tumors could be malignant.[92][45] Despite the differences between these two original categories, and further complexities due to subsequent inclusion of other NETs of pancreas and pulmonary origin, all NETs are sometimes (incorrectly) subsumed into the term "carcinoid".
Enterochromaffin cells, which give rise to carcinoid tumors, were identified in 1897 by Nikolai Kulchitsky and their secretion of serotonin was established in 1953[92] when the "flushing" effect of serotonin had become clinically recognized. Carcinoid heart disease was identified in 1952, and carcinoid fibrosis in 1961.[92]
Neuroendocrine tumors were sometimes called APUDomas because these cells often show amine precursor (L-DOPA and 5-hydroxytryptophan) uptake and decarboxylation to produce biogenic amines such as catecholamines and serotonin. Although this behavior was also part of the disproven hypothesis that these cells might all embryologically arise from the neural crest,[56][66][67] neuroendocrine cells sometimes produce various types of hormones and amines,[67] and they can also have strong receptors for other hormones to which they respond.
There have been multiple nomenclature systems for these tumors,[2] and the differences between these schema have often been confusing. Nonetheless, these systems all distinguish between well-differentiated (low and intermediate-grade) and poorly differentiated (high-grade) NETs. Cellular proliferative rate is of considerable significance in this prognostic assessment.[2]
References
- Ramage JK, Davies AH, Ardill J, et al. (June 2005). "Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours". Gut. 54. 54 (Suppl 4): iv1–iv16. doi:10.1136/gut.2004.053314. PMC 1867801. PMID 15888809. Archived from the original on 2008-12-09.
- Klimstra, D.S.; Modlin, I.R.; Coppola, D.; Lloyd, R.V.; Suster, S. (2010). "The Pathologic Classification of Neuroendocrine Tumors". Pancreas. 39 (6): 707–12. doi:10.1097/MPA.0b013e3181ec124e. PMID 20664470.
- Tan, E.H.; Tan, C. (2011). "Imaging of gastroenteropancreatic neuroendocrine tumors". World Journal of Clinical Oncology. 2 (1): 28–43. doi:10.5306/wjco.v2.i1.28. PMC 3095463. PMID 21603312.
- Van Eeden, S.; Offerhaus, G.J.A.; Hart, A.A.M.; Boerrigter, L.; Nederlof, P.M.; Porter, E.; Van Velthuysen, M.L.F. (2007). "Goblet cell carcinoid of the appendix: A specific type of carcinoma". Histopathology. 51 (6): 763–73. doi:10.1111/j.1365-2559.2007.02883.x. PMID 18042066.
- Liu Y, Sturgis CD, Grzybicki DM, et al. (September 2001). "Microtubule-associated protein-2: a new sensitive and specific marker for pulmonary carcinoid tumor and small cell carcinoma". Mod. Pathol. 14 (9): 880–85. doi:10.1038/modpathol.3880406. PMID 11557784.
- Rufini V, Calcagni ML, Baum RP (July 2006). "Imaging of neuroendocrine tumors". Semin Nucl Med. 36 (3): 228–47. doi:10.1053/j.semnuclmed.2006.03.007. PMID 16762613.
- Soga, J. (2003). "Carcinoids and their variant endocrinomas. An analysis of 11842 reported cases". Journal of Experimental & Clinical Cancer Research. 22 (4): 517–30. PMID 15053292.
- Soga, J.; Yakuwa, Y.; Osaka, M. (1999). "Evaluation of 342 cases of mediastinal/thymic carcinoids collected from literature: A comparative study between typical carcinoids and atypical varieties". Annals of Thoracic and Cardiovascular Surgery. 5 (5): 285–92. PMID 10550713.
- Oberg, K.; Jelic, S.; Esmo Guidelines Working, G. (2008). "Neuroendocrine bronchial and thymic tumors: ESMO Clinical Recommendation for diagnosis, treatment and follow-up". Annals of Oncology. 19: ii102–ii103. doi:10.1093/annonc/mdn116. PMID 18456740.
- Beasley, M.; Brambilla, E.; Travis, W. (2005). "The 2004 World Health Organization classification of lung tumors". Seminars in Roentgenology. 40 (2): 90–97. doi:10.1053/j.ro.2005.01.001. PMID 15898407.
- Gustafsson, B.I.; Kidd, M.; Chan, A.; Malfertheiner, M.V.; Modlin, I.M. (2008). "Bronchopulmonary neuroendocrine tumors". Cancer. 113 (1): 5–21. doi:10.1002/cncr.23542. PMID 18473355.
- Wick, M.; Berg, L.; Hertz, M. (1992). "Large cell carcinoma of the lung with neuroendocrine differentiation. A comparison with large cell "undifferentiated" pulmonary tumors". American Journal of Clinical Pathology. 97 (6): 796–805. doi:10.1093/ajcp/97.6.796. PMID 1317668.
- Massironi, S.; Sciola, V.; Peracchi, M.; Ciafardini, C.; Spampatti, M.; Conte, D. (2008). "Neuroendocrine tumors of the gastro-entero-pancreatic system". World Journal of Gastroenterology. 14 (35): 5377–84. doi:10.3748/wjg.14.5377. PMC 2744160. PMID 18803349.
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; De Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle Fave, G.; Kaltsas, G.A.; Krenning, E.P.; Moss, S.F.; Nilsson, O.; Rindi, G.; Salazar, R.; Ruszniewski, P.; Sundin, A. (2008). "Gastroenteropancreatic neuroendocrine tumours". The Lancet Oncology. 9 (1): 61–72. doi:10.1016/S1470-2045(07)70410-2. PMID 18177818.
- Metz, D.C.; Jensen, R.T. (2008). "Gastrointestinal Neuroendocrine Tumors: Pancreatic Endocrine Tumors". Gastroenterology. 135 (5): 1469–92. doi:10.1053/j.gastro.2008.05.047. PMC 2612755. PMID 18703061.
- Griniatsos, J.; Michail, O. (2010). "Appendiceal neuroendocrine tumors: Recent insights and clinical implications". World Journal of Gastrointestinal Oncology. 2 (4): 192–96. doi:10.4251/wjgo.v2.i4.192. PMC 2999180. PMID 21160597.
- Ni, S.; Sheng, W.; Du, X. (2010). "Pathologic research update of colorectal neuroendocrine tumors". World Journal of Gastroenterology. 16 (14): 1713–19. doi:10.3748/wjg.v16.i14.1713. PMC 2852818. PMID 20380002.
- Konishi, T.; Watanabe, T.; Nagawa, H.; Oya, M.; Ueno, M.; Kuroyanagi, H.; Fujimoto, Y.; Akiyoshi, T.; Yamaguchi, T.; Muto, T. (2010). "Treatment of colorectal carcinoids: A new paradigm". World Journal of Gastrointestinal Surgery. 2 (5): 153–56. doi:10.4240/wjgs.v2.i5.153. PMC 2999232. PMID 21160865.
- Soga, J. (2002). "Primary hepatic endocrinomas (carcinoids and variant neoplasms). A statistical evaluation of 126 reported cases". Journal of Experimental & Clinical Cancer Research. 21 (4): 457–68. PMID 12636090.
- C., C.; M., N.; V., M. (2004). "Primary hepatic carcinoid tumours". HPB. 6 (1): 13–17. doi:10.1080/13651820310017228. PMC 2020649. PMID 18333038.
- Moriura, S.; Ikeda, S.; Hirai, M.; Naiki, K.; Fujioka, T.; Yokochi, K.; Gotou, S. (1993). "Hepatic gastronoma". Cancer. 72 (5): 1547–50. doi:10.1002/1097-0142(19930901)72:5<1547::AID-CNCR2820720510>3.0.CO;2-C. PMID 8348490.
- Soga, J. (2003). "Primary endocrinomas (carcinoids and variant neoplasms) of the gallbladder. A statistical evaluation of 138 reported cases". Journal of Experimental & Clinical Cancer Research. 22 (1): 5–15. PMID 12725316.
- Soga, J.; Osaka, M.; Yakuwa, Y. (2001). "Gut-endocrinomas (carcinoids and related endocrine variants) of the breast: An analysis of 310 reported cases". International Surgery. 86 (1): 26–32. PMID 11890336.
- Murali, R.; Kneale, K.; Lalak, N.; Delprado, W. (2006). "Carcinoid tumors of the urinary tract and prostate". Archives of Pathology & Laboratory Medicine. 130 (11): 1693–1706. doi:10.1043/1543-2165(2006)130[1693:CTOTUT]2.0.CO;2 (inactive 2019-12-03). ISSN 1543-2165. PMID 17076534.
- Mikuz, G. (1993). "Non-urothelial tumors of the urinary tract". Verhandlungen der Deutschen Gesellschaft für Pathologie. 77: 180–98. PMID 7511278.
- Soga, J.; Osaka, M.; Yakuwa, Y. (2001). "Gut-endocrinomas (carcinoids and related endocrine variants) of the uterine cervix: An analysis of 205 reported cases". Journal of Experimental & Clinical Cancer Research. 20 (3): 327–34. PMID 11718210.
- Usmani, S; Orevi, M; Stefanelli, A; Zaniboni, A; Gofrit, ON; Bnà, C; Illuminati, S; Lojacono, G; Noventa, S; Savelli, G (June 2019). "Neuroendocrine differentiation in castration resistant prostate cancer. Nuclear medicine radiopharmaceuticals and imaging techniques: A narrative review". Critical Reviews in Oncology/Hematology. 138: 29–37. doi:10.1016/j.critrevonc.2019.03.005. PMID 31092382.
- Davies, AH; Beltran, H; Zoubeidi, A (May 2018). "Cellular plasticity and the neuroendocrine phenotype in prostate cancer". Nature Reviews. Urology. 15 (5): 271–286. doi:10.1038/nrurol.2018.22. PMID 29460922.
- Jensen, R.T.; Berna, M.J.; Bingham, D.B.; Norton, J.A. (2008). "Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies". Cancer. 113 (7 Suppl): 1807–43. doi:10.1002/cncr.23648. PMC 2574000. PMID 18798544.
- Hirsch, N.P.; Murphy, A.; Radcliffe, J. (2001). "Neurofibromatosis: Clinical presentations and anaesthetic implications". British Journal of Anaesthesia. 86 (4): 555–64. doi:10.1093/bja/86.4.555. PMID 11573632.
- Lodish, M.B.; Stratakis, C.A. (2010). "Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes". Best Practice & Research Clinical Endocrinology & Metabolism. 24 (3): 439–49. doi:10.1016/j.beem.2010.02.002. PMC 2939061. PMID 20833335.
- Dworakowska, D.; Grossman, A.B. (2008). "Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review". Endocrine-Related Cancer. 16 (1): 45–58. doi:10.1677/ERC-08-0142. PMID 18978035.
- OMIM - Online Mendelian Inheritance in Man. Carney Complex, type 1; CNC1 (OMIM 160980) omim.org
- OMIM—Online Mendelian Inheritance in Man. Carney Complex, type 2; CNC2 (OMIM 605244)
- >Bosman, Fred T.; Carneiro, Fatima; Hruban, Ralph H.; Theise, Neil D., eds. (2010). WHO Classification of Tumours of the Digestive System (4 ed.). Lyon: International Agency for Research on Cancer. pp. 13–14. ISBN 978-92-832-2432-7.
- Basturk, O; Yang, Z; Tang, LH; Hruban, RH; Adsay, V; McCall, CM; Krasinskas, AM; Jang, KT; Frankel, WL; Balci, S; Sigel, C; Klimstra, DS (2015). "The high-grade (WHO G3) pancreatic neuroendocrine tumor category is morphologically and biologically heterogenous and includes both well differentiated and poorly differentiated neoplasms". The American Journal of Surgical Pathology. 39 (5): 683–90. doi:10.1097/PAS.0000000000000408. PMC 4398606. PMID 25723112.
- Amin, Mahul B., ed. (2017). AJCC Cancer Staging Manual (8 ed.). Springer. p. 351. ISBN 978-3-319-40617-6.
- Amin, Mahul B., ed. (2017). "29 - Neuroendocrine Tumors of the Stomach". AJCC Cancer Staging Manual (8 ed.). Springer. p. 355. ISBN 978-3-319-40617-6.
- Amin, Mahul B., ed. (2017). "30 - Neuroendocrine Tumors of the Duodenum & Ampulla of Vater". AJCC Cancer Staging Manual (8 ed.). Springer. p. 369. ISBN 978-3-319-40617-6.
- Amin, Mahul B., ed. (2017). "31 - Neuroendocrine Tumors of the Jejunum and Ileum". AJCC Cancer Staging Manual (8 ed.). Springer. pp. 379–380. ISBN 978-3-319-40617-6.
- Amin, Mahul B., ed. (2017). "32 - Neuroendocrine Tumors of the Appendix". AJCC Cancer Staging Manual (8 ed.). Springer. p. 392. ISBN 978-3-319-40617-6.
- Amin, Mahul B., ed. (2017). "33 - Neuroendocrine Tumors of the Colon and Rectum". AJCC Cancer Staging Manual (8 ed.). Springer. p. 399. ISBN 978-3-319-40617-6.
- Amin, Mahul B., ed. (2017). "30 - Neuroendocrine Tumors of the Pancreas". AJCC Cancer Staging Manual (8 ed.). Springer. pp. 415–416. ISBN 978-3-319-40617-6.
- Kimura W, Kuroda A, Morioka Y (July 1991). "Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases". Dig. Dis. Sci. 36 (7): 933–42. doi:10.1007/BF01297144. PMID 2070707. "[In] 800 autopsy cases, ... incidence of tumor was 10% (6/60) in individuals having histiological studies of all sections of the pancreas"
- Arnold R, Göke R, Wied M, Behr T (2003). "Chapter 15 Neuroendocrine Gastro-Entero-Pancreatic (GEP) Tumors". In Scheppach W, Bresalier RS, Tytgat GN (eds.). Gastrointestinal and Liver Tumors. Berlin: Springer. pp. 195–233. ISBN 978-3-540-43462-7.
- Pommier R. 2003. The role of surgery and chemoembolization in the management of carcinoid. California Carcinoid Fighters Conference. October 25, carcinoid.org Archived 2015-09-15 at the Wayback Machine
- Health Communities. Carcinoid Tumor Overview. healthcommunities.com Archived 2012-03-03 at the Wayback Machine
- Kvols LK. 2002. Carcinoid Tumors and the Carcinoid Syndrome: What's New in the Therapeutic Pipeline. (The Carcinoid Cancer Foundation: Carcinoid Symposium 2002) carcinoid.org Archived 2015-01-05 at the Wayback Machine
- Benson AB, Myerson RJ, and Sasson AR. Pancreatic, neuroendocrine GI, and adrenal cancers. Cancer Management: A Multidisciplinary Approach 13th edition 2010; ISBN 978-0-615-41824-7 Text is available electronically (but may require free registration) at Infosite Archived 2011-05-15 at the Wayback Machine, cancernetwork.com; accessed November 8, 2015.
- Pancreatic Neuroendocrine Tumors (Islet Cell Tumors) Treatment (PDQ) Health Professional Version. National Cancer Institute. March 7, 2014. ncbi.nlm.nih.gov
- Burns WR, Edil BH (March 2012). "Neuroendocrine pancreatic tumors: guidelines for management and update". Current Treatment Options in Oncology. 13 (1): 24–34. doi:10.1007/s11864-011-0172-2. PMID 22198808.
- Klimstra DS, Modlin IR, Coppola D, et al. (August 2010). "The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems" (PDF). Pancreas. 39 (6): 707–12. doi:10.1097/MPA.0b013e3181ec124e. PMID 20664470.
- Bostanci A (2005). "Wildlife biology. A devil of a disease". Science. 307 (5712): 1035. doi:10.1126/science.307.5712.1035. PMID 15718445.
The tumors [of Devil facial tumor disease] have been characterized as a neuroendocrine cancer
- Kinver, Mark (January 1, 2010). "Tasmanian devil facial cancer origins 'identified'". BBC. Archived from the original on January 2, 2010.
- Walsh, Bryan (January 1, 2010). "Decoding the Tasmanian Devil's Deadly Cancer". Time. Archived from the original on January 8, 2010.
- Langley, K. (1994). "The Neuroendocrine Concept Today". Annals of the New York Academy of Sciences. 733 (1): 1–17. Bibcode:1994NYASA.733....1L. doi:10.1111/j.1749-6632.1994.tb17251.x. PMID 7978856.
- Ferolla, P.; Faggiano, A.; Mansueto, G.; Avenia, N.; Cantelmi, M.; Giovenali, P.; Del Basso De Caro ML; Milone, F.; Scarpelli, G.; Masone, S.; Santeusanio, F.; Lombardi, G.; Angeletti, G.; Colao, A. (2008). "The biological characterization of neuroendocrine tumors: The role of neuroendocrine markers". Journal of Endocrinological Investigation. 31 (3): 277–86. doi:10.1007/bf03345602. PMID 18401212.
- Prince, Jim McMorran, Damian Crowther, Stew McMorran, Steve Youngmin, Ian Wacogne, Jon Pleat, Clive. "investigations - General Practice Notebook". www.gpnotebook.co.uk. Archived from the original on 2017-02-24. Retrieved 2017-02-23.
- Oberg, K (July 2005). "Neuroendocrine tumors of the gastrointestinal tract: recent advances in molecular genetics, diagnosis, and treatment". Current Opinion in Oncology. 17 (4): 386–91. doi:10.1097/01.cco.0000167739.56948.a9. PMID 15933475.
- Hofman, M.S.; Kong, G.; Neels, O.C.; Eu, P.; Hong, E.; Hicks, R.J. (2012). "High management impact of Ga-68 DOTATATE (GaTate) PET/CT for imaging neuroendocrine and other somatostatin expressing tumours". Journal of Medical Imaging and Radiation Oncology. 56 (1): 40–47. doi:10.1111/j.1754-9485.2011.02327.x. PMID 22339744.
- Hofman, M.S.; Hicks, R.J. (2012). "Changing paradigms with molecular imaging of neuroendocrine tumors". Discovery Medicine. 14 (74): 71–81. PMID 22846204. Retrieved November 8, 2015.
- Nilica, Bernhard; Waitz, Dietmar; Stevanovic, Vlado; Uprimny, Christian; Kendler, Dorota; Buxbaum, Sabine; Warwitz, Boris; Gerardo, Llanos; Henninger, Benjamin (2016-08-01). "Direct comparison of 68Ga-DOTA-TOC and 18F-FDG PET/CT in the follow-up of patients with neuroendocrine tumour treated with the first full peptide receptor radionuclide therapy cycle". European Journal of Nuclear Medicine and Molecular Imaging. 43 (9): 1585–1592. doi:10.1007/s00259-016-3328-2. ISSN 1619-7070. PMC 4932132. PMID 26922350.
- Berretta, M. (2010). "Biomarkers in neuroendocrine tumors". Frontiers in Bioscience. S2: 332–42. doi:10.2741/s68. PMID 20036951.
- Mukhopadhyay S, Dermawan JK, Lanigan CP, Farver CF (August 2018). "Insulinoma-associated protein 1 (INSM1) is a sensitive and highly specific marker of neuroendocrine differentiation in primary lung neoplasms: an immunohistochemical study of 345 cases, including 292 whole-tissue sections". Modern Pathology. Epub ahead of print (1): 100–109. doi:10.1038/s41379-018-0122-7. PMID 30154579.
- Cameron K Tebbi, MD; Chief Editor: Max J Coppes, MD, PhD, MBA; et al. (1 Apr 2014). "Carcinoid Tumor". Medscape.com. WebMD LLC. Archived from the original on 15 December 2014. Retrieved 3 September 2014.CS1 maint: multiple names: authors list (link)
- Warner, R.R.P. (2005). "Enteroendocrine Tumors Other Than Carcinoid: A Review of Clinically Significant Advances". Gastroenterology. 128 (6): 1668–84. doi:10.1053/j.gastro.2005.03.078. PMID 15887158.
- Öberg, K. (1998). "Carcinoid Tumors: Current Concepts in Diagnosis and Treatment". The Oncologist. 3 (5): 339–45. PMID 10388123.
- Oberg, K.; Akerstrom, G.; Rindi, G.; Jelic, S.; Esmo Guidelines Working, G. (2010). "Neuroendocrine gastroenteropancreatic tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up". Annals of Oncology. 21: v223–v227. doi:10.1093/annonc/mdq192. PMID 20555086.
- nccn.org
- National Cancer Institute. Islet Cell Tumors (Endocrine Pancreas) cancer.gov Archived 2011-06-07 at the Wayback Machine
- National Cancer Institute. Gastrointestinal Carcinoid Tumors Treatment cancer.gov Archived 2011-06-27 at the Wayback Machine
- National Cancer Institute. Merkel cell tumors, cancer.gov Archived 2011-06-07 at the Wayback Machine
- National Cancer Institute. Pheochromocytoma and Paraganglioma cancer.gov Archived 2011-06-07 at the Wayback Machine
- "Neuroendocrine tumors, NCCN Guidelines Version 1.2015" (PDF). NCCN Guidelines. National Comprehensive Cancer Network, Inc. November 11, 2014. Retrieved December 25, 2014.
- Oberg, K.; Kvols, L.; Caplin, M.; Delle Fave, G.; De Herder, W.; Rindi, G.; Ruszniewski, P.; Woltering, E.; Wiedenmann, B. (2004). "Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system". Annals of Oncology. 15 (6): 966–73. doi:10.1093/annonc/mdh216. PMID 15151956.
- asco.org Archived 2012-03-23 at the Wayback Machine; accessed November 8, 2015.
- Caplin ME, Pavel M, Cwikla JB, et al. (July 17, 2014). "Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors". The New England Journal of Medicine. 371 (3): 224–33. doi:10.1056/NEJMoa1316158. PMID 25014687.
- Öberg K. Neuroendocrine Gastroenteropancreatic Tumours: Current Views on Diagnosis and Treatment. Business Briefing. European Oncology Review 2005; pp. 1–6.
- Strosberg, Jonathan; El-Haddad, Ghassan; Wolin, Edward; Hendifar, Andrew; Yao, James; Chasen, Beth; Mittra, Erik; Kunz, Pamela L.; Kulke, Matthew H. (2017-01-11). "Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors". New England Journal of Medicine. 376 (2): 125–135. doi:10.1056/nejmoa1607427. hdl:2445/125256. PMC 5895095. PMID 28076709.
- Kratochwil, C.; Giesel, F. L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. (2014-11-01). "213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: a first-in-human experience". European Journal of Nuclear Medicine and Molecular Imaging. 41 (11): 2106–2119. doi:10.1007/s00259-014-2857-9. ISSN 1619-7070. PMC 4525192. PMID 25070685.
- Kashyap, R; Hofman, M. S.; Michael, M; Kong, G; Akhurst, T; Eu, P; Zannino, D; Hicks, R. J. (2015). "Favourable outcomes of (177)Lu-octreotate peptide receptor chemoradionuclide therapy in patients with FDG-avid neuroendocrine tumours". European Journal of Nuclear Medicine and Molecular Imaging. 42 (2): 176–85. doi:10.1007/s00259-014-2906-4. PMID 25209134.
- Hofman, M. S.; Michael, M; Kashyap, R; Hicks, R. J. (2015). "Modifying the Poor Prognosis Associated with 18F-FDG-Avid NET with Peptide Receptor Chemo-Radionuclide Therapy (PRCRT)". Journal of Nuclear Medicine. 56 (6): 968–9. doi:10.2967/jnumed.115.154500. PMID 25814516.
- Fong, T and Schoenfield LJ. Arterial Chemotherapy Infusion of the Liver (and) Chemoembolization of the Liver (TACE) medicinenet.com Archived 2014-12-24 at the Wayback Machine; accessed November 8, 2015.
- Welsh, J.; Kennedy, A.; Thomadsen, B. (2006). "Selective internal radiation therapy (SIRT) for liver metastases secondary to colorectal adenocarcinoma". International Journal of Radiation OncologyBiologyPhysics. 66 (2): S62–S73. doi:10.1016/j.ijrobp.2005.09.011. PMID 16979443.
- Van De Wiele, C.; Defreyne, L.; Peeters, M.; Lambert, B. (2009). "Yttrium-90 labelled resin microspheres for treatment of primary and secondary malignant liver tumors". The Quarterly Journal of Nuclear Medicine and Molecular Imaging. 53 (3): 317–24. PMID 19521311.
- Salem, R.; Thurston, K.; Carr, B.; Goin, J.; Geschwind, J. (2002). "Yttrium-90 microspheres: Radiation therapy for unresectable liver cancer". Journal of Vascular and Interventional Radiology. 13 (9 Pt 2): S223–S229. doi:10.1016/S1051-0443(07)61790-4. PMID 12354840.
- Masters, Alexander (2014-10-14). "A plutocratic proposal". Mosaic. The Wellcome Trust. Archived from the original on 2016-05-29. Retrieved 2016-07-03.
- "iCancer web site". icancer.org.uk. Archived from the original on 2016-07-14. Retrieved 2016-07-03.
- Masters, Alexander (2016-07-02). "Can crowdfunding really cure cancer? Alexander Masters investigates a pioneering new project". The Telegraph. Archived from the original on 2016-07-03. Retrieved 2016-07-03.
- Öberg, K.; Castellano, D. (2011). "Current knowledge on diagnosis and staging of neuroendocrine tumors". Cancer and Metastasis Reviews. 30: 3–7. doi:10.1007/s10555-011-9292-1. PMID 21311954.
- Kimura, W.; Kuroda, A.; Morioka, Y. (1991). "Clinical pathology of endocrine tumors of the pancreas". Digestive Diseases and Sciences. 36 (7): 933–42. doi:10.1007/BF01297144. PMID 2070707.
- Modlin, I.M.; Shapiro, M.D.; Kidd, M. (2004). "Siegfried oberndorfer: Origins and perspectives of carcinoid tumors". Human Pathology. 35 (12): 1440–51. doi:10.1016/j.humpath.2004.09.018. PMID 15619202.