Medrogestone
Medrogestone, sold under the brand name Colprone among others, is a progestin medication which has been used in menopausal hormone therapy and in the treatment of gynecological disorders.[6][2] It is available both alone and in combination with an estrogen.[7] It is taken by mouth.[2][8]
 | |
| Clinical data | |
|---|---|
| Trade names | Colprone, others |
| Other names | Metrogestone; Medrogesterone; AY-62022, NSC-123018, R-13615; 6,17α-Dimethyl-6-dehydroprogesterone; 6,17α-Dimethyl-4,6-pregnadiene-3,20-dione |
| Pregnancy category |
|
| Routes of administration | By mouth |
| Drug class | Progestogen; Progestin |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | Nearly 100%[1][2] |
| Protein binding | 95%: Albumin (90%), CBG (3%), SHBG (2%)[2] |
| Metabolism | Hepatic (hydroxylation)[1] |
| Elimination half-life | 35–36 hours[3][4][5] |
| Identifiers | |
IUPAC name
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.012.323 |
| Chemical and physical data | |
| Formula | C23H32O2 |
| Molar mass | 340.507 g/mol g·mol−1 |
| 3D model (JSmol) | |
SMILES
| |
InChI
| |
Medrogestone is a progestin, or a synthetic progestogen, and hence is an agonist of the progesterone receptor, the biological target of progestogens like progesterone.[2] It has weak antiandrogenic, glucocorticoid, and antimineralocorticoid activity and no other important hormonal activity.[2][1][9][10] Due to its progestogenic activity, medrogestone has antigonadotropic effects.[1][2]
Medrogestone was described as early as 1963 and was introduced for medical use by at least 1966.[11][12][9] It has mostly been discontinued and remains available only in a few countries.[13][7]
Medical uses
In the past, medrogestone was used in the treatment of endometrial cancer and in some regimens for breast cancer, and, in men, for benign prostatic hyperplasia. It still finds use in the treatment of amenorrhea[14] and as the progestin component in certain forms of menopausal hormone therapy.[15]
Cyclic treatment with low-dose (10 mg/day) medrogestone has been found to be effective in the treatment of fibrocystic breast changes and associated mastodynia (breast pain).[16]
Contraindications
Intrahepatic cholestasis of pregnancy (acute or in history), vaginal bleeding of unknown origin, and severe diseases of the liver such as tumors are absolute contraindications for medrogestone, as are thrombotic events such as thrombophlebitis or stroke.[17] Relative contraindications include a history of jaundice or itching in pregnancy or gestational pemphigoid.
Medrogestone is contraindicated during pregnancy because progestogens are associated with risks for the fetus in animals and humans.[18] Studies in pregnant rabbits have shown skeletal deformations under 3 mg/kg medrogestone but not under 1 mg/kg. Typical therapeutic doses of the drug are between 0.1 and 0.25 mg/kg.
It is not known whether medrogestone passes into breast milk, but it is to be expected given its lipophilicity and studies with structurally related progestins.[18]
Side effects
Medrogestone seldom produces side effects, all of which are typical of progestogens. They include nausea, depression,[17] decreased appetite, headache, and dizziness.
Overdose
The acute toxicity of the drug is low. Overdose causes only harmless side effects such as nausea and vaginal bleeding.[18] The LD50 has been found to range between 500 mg/kg in dogs and over 3000 mg/kg in rats. Chronic toxicity has been examined in animals, but nothing but the typical adverse effects of progestogens, and reduction of prostatic weight in rhesus monkeys, have been found. Accidental intake of the drug, including in children, is normally not dangerous. Intake of extremely large doses, or intake by patients with epilepsy or impaired kidney function, can result in central nervous cramping.[17]
Interactions
Enzyme inducers such as barbiturates, phenylbutazone, phenytoin, ampicillin or tetracyclines are expected to reduce plasma concentrations of medrogestone, but no systematic research has been done.[18]
Pharmacology
Pharmacodynamics
Medrogestone is described as a pure progestogen similar in profile to progesterone.[9][19] In contrast to progesterone however, medrogestone is more potent and is orally active.[9] There is reportedly no information available on the receptor binding of medrogestone at the various steroid hormone receptors.[2] However, based on animal research (e.g., the Clauberg test and other assays), medrogestone appears to be a potent progestogen, devoid of androgenic, estrogenic, and glucocorticoid activity, but with weak antiandrogenic and very weak antimineralocorticoid activity.[9] Accordingly, no evidence of androgenic or glucocorticoid activity, including effects on the estrogen-induced increase in triglycerides and HDL cholesterol and adrenal suppression, were observed in clinical studies.[2][20] However, in a very high-dosage (100 mg/day for 6 months) study of medrogestone for benign prostatic hyperplasia, a hyperglycemic effect and changes in plasma cortisol levels were observed and considered likely to be secondary to glucocorticoid activity, and decreased sodium levels were also observed and attributed to antimineralocorticoid activity.[10] In any case, under normal circumstances (i.e., at typical clinical dosages), medrogestone is described as a progestogen and antigonadotropin and weak antiandrogen in humans without other clinically relevant activity.[1][2]
Medrogestone has been found to be an inhibitor of 3β-hydroxysteroid dehydrogenase/Δ5-4 isomerase in vitro, preventing conversion of pregnenolone to progesterone and 17α-hydroxypregnenolone to 17α-hydroxyprogesterone in rat testis preparations, and inhibits the biosynthesis of testosterone in vivo in rats.[21][22][23] In addition, similarly to progesterone, medrogestone can inhibit 5α-reductase in vitro in microsomal preparations of skin and prostate.[24] Although their clinical relevance is uncertain, these actions of medrogestone could contribute to its weak antiandrogen activity.[25]
Pharmacokinetics
Upon oral administration, medrogestone is rapidly absorbed, and the bioavailability is nearly 100%.[1][2] After ingestion of a 10 mg dose of medrogestone, peak circulating concentrations (Cmax) of 10–15 ng/mL are achieved.[2] The distribution and elimination half-lives of medrogestone are 4 hours and 35–36 hours, respectively.[2][3][4] The drug is largely bound (90%) to albumin, and to only small extents to corticosteroid-binding globulin (3%) and sex hormone-binding globulin (2%).[2] The metabolism of medrogestone is most importantly by hydroxylation.[1][2]
Chemistry
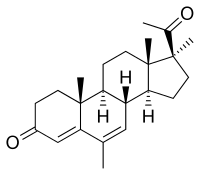
Medrogestone, also known as 6,17α-dimethyl-6-dehydroprogesterone or as 6,17α-dimethyl-4,6-pregnadiene-3,20-dione, is a synthetic pregnane steroid and a derivative of progesterone. It is structurally related to the 17α-hydroxyprogesterone derivatives megestrol acetate and medroxyprogesterone acetate.[1][2] Medrogestone itself is not a 17α-hydroxyprogesterone derivative and is instead a derivative of 17α-methylprogesterone.[1][2] This is because it features a methyl group at the C17α position instead of a hydroxy or acetoxy group.[1][2] In addition to its C17α methyl group, medrogestone possesses a methyl group at the C6 position and a double bond between the C6 and C7 positions.[1] The only structural difference between medrogestone and megestrol acetate is the replacement of the C17α acetoxy group with a methyl group.[11][13][7]
Synthesis
The oral activity of 17α-methylprogesterone has already been alluded to. This compound, which may well owe this property to the inhibition of metabolism in a manner analogous to synthetic androgens and estrogens, is not sufficiently potent in its own right to constitute a useful drug. Incorporation of known potentiating modifications yields the commercially available oral progestin medrogestone (4).
The preparation of the 6-methyl-16-dehydropregnenolone acetate (1) precursor is covered here.
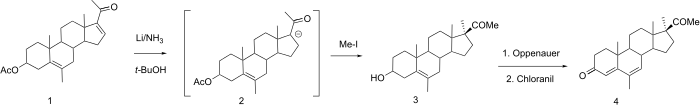
Reduction of the conjugated 16,17 double bond of 6-methyl-16-dehydropregnenolone acetate by means of lithium in liquid ammonia leads initially to the 17 enolate ion; this is alkylated in situ with methyl iodide. The now-familiar steric control asserts itself to afford the 17α-methyl compound,.
The acetate group is lost as a side reaction. In an interesting modification on the usual scheme, (3) is treated with aluminum isopropoxide and a ketone (Oppenauer conditions) as well as chloranil in a single reaction; the 4,6-diene, (medrogesterone), is obtained directly from this step.
History
Medrogestone was first described as early as 1963 and appears to have been marketed since at least 1966.[11][12][9]
Society and culture
Generic names
Medrogestone is the generic name of the drug and its INN, USAN, and BAN.[11][13][6][7] It is also known by its developmental code names AY-62022, NSC-123018, and R-13615.[11][13][6][7]
Brand names
Medrogestone is or has been marketed under the brand names Ayerluton, Colpro, Colpron, Colprone, Etogyn, Prothil, and, in combination with conjugated estrogens, Presomen.[11][13][7]
Availability
Medrogestone has been marketed in the United States[9] and Canada and widely throughout Europe, as well as in Argentina, Hong Kong, and other countries.[13] However, it is no longer available in the United States[27] or many other countries, and is reported to remain marketed only in a few countries including France, Germany, Tunisia, and Egypt.[7]
References
- Schindler AE, Campagnoli C, Druckmann R, Huber J, Pasqualini JR, Schweppe KW, Thijssen JH (December 2003). "Classification and pharmacology of progestins" (PDF). Maturitas. 46 Suppl 1: S7–S16. doi:10.1016/j.maturitas.2003.09.014. PMID 14670641.
- Kuhl H (2005). "Pharmacology of estrogens and progestogens: influence of different routes of administration" (PDF). Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947.
- Stanczyk FZ (2002). "Pharmacokinetics and potency of progestins used for hormone replacement therapy and contraception". Reviews in Endocrine & Metabolic Disorders. 3 (3): 211–24. doi:10.1023/A:1020072325818. PMID 12215716.
Medrogestone The pharmacokinetics of medrogestone (5 mg dose) was studied in 12 Chinese young males who received a single oral dose of this drug [20]. The mean ± standard deviation Cmax was 8.21 ± 2.78 ng/ml and Tmax was 2.57 ± 1.02; the half-life of elimination was 34.9 ± 17.0 hours.
- Lobo R, Crosignani P, Paoletti R, Bruschi F (6 December 2012). Women’s Health and Menopause: New Strategies — Improved Quality of Life. Springer Science & Business Media. pp. 142–. ISBN 978-1-4615-1061-1.
- Stanczyk FZ, Hapgood JP, Winer S, Mishell DR (April 2013). "Progestogens used in postmenopausal hormone therapy: differences in their pharmacological properties, intracellular actions, and clinical effects". Endocrine Reviews. 34 (2): 171–208. doi:10.1210/er.2012-1008. PMC 3610676. PMID 23238854.
- Morton I, Morton I, Hall JM (31 October 1999). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 173–. ISBN 978-0-7514-0499-9.
- https://www.drugs.com/international/medrogestone.html
- Winnifred Berg Cutler; Celso-Ramon Garcia (1993). Menopause: A Guide for Women and Those who Love Them. Norton. pp. 134–. ISBN 978-0-393-30995-9.
- Revesz C, Chappel CI (December 1966). "Biological activity of medrogestone: a new orally active progestin". Journal of Reproduction and Fertility. 12 (3): 473–87. doi:10.1530/jrf.0.0120473. PMID 4288903.
- Rangno RE, McLeod PJ, Ruedy J, Ogilvie RI (1971). "Treatment of benign prostatic hypertrophy with medrogestone". Clinical Pharmacology and Therapeutics. 12 (4): 658–65. doi:10.1002/cpt1971124658. PMID 4105445.
- Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 759–760. ISBN 978-1-4757-2085-3.
- Deghenghi, R.; Lefebvre, Y.; Mitchell, P.; Morand, P.F.; Gaudry, R. (1963). "The synthesis of 17α-methylprogesterone derivatives". Tetrahedron. 19 (2): 289–298. doi:10.1016/S0040-4020(01)98530-8. ISSN 0040-4020.
- Swiss Pharmaceutical Society (January 2000). Index Nominum 2000: International Drug Directory. Taylor & Francis. pp. 638–. ISBN 978-3-88763-075-1.
- Medrogestone at the US National Library of Medicine Medical Subject Headings (MeSH)
- Sweetman SC, ed. (2009). "Sex hormones and their modulators". Martindale: the complete drug reference (36th ed.). London: Pharmaceutical Press. p. 2113. ISBN 978-0-85369-840-1.
- Winkler UH, Schindler AE, Brinkmann US, Ebert C, Oberhoff C (2001). "Cyclic progestin therapy for the management of mastopathy and mastodynia". Gynecol. Endocrinol. 15 Suppl 6: 37–43. doi:10.1080/gye.15.s6.37.43. PMID 12227885.
- Pittner H, Jasek W, Wicho H, eds. (2006). Austria-Codex (in German). 1 (2006/2007 ed.). Vienna: Österreichischer Apothekerverlag. p. 1696. ISBN 978-3-85200-177-7.
- "Fachinformation zu Colpro" [Colpro summary of product characteristics] (in German). Open Drug Database. November 1997. Retrieved 15 August 2010.
- Gautam Allahbadia (29 February 2016). Manual of Ovulation Induction & Ovarian Stimulation Protocols. JP Medical Ltd. pp. 94–. ISBN 978-93-5090-958-4.
The natural preparations of P4 include progesterone, dydrogesterone, and medrogestone.
- Carter, W. F.; Faucher, G. L.; Greenblatt, R. B. (1964). "Evaluation of a new progestational agent, 6, 17α-dimethyl-6-dehydroprogesterone". American Journal of Obstetrics and Gynecology. 89 (5): 635–41. doi:10.1016/0002-9378(64)90158-9. PMID 14176912.
- Givner, M. L.; Dvornik, D. (1972). "Inhibition of testosterone biosynthesis by medrogestone". Experientia. 28 (9): 1105–1106. doi:10.1007/BF01918702. ISSN 0014-4754. PMID 4269193.
In vitro, medrogestone blocks the synthesis of hormones in the gonads through inhibition of 3β-hydroxysteroid Δ4,5 isomerase. In vivo, medrogestone reduced testosterone levels in the rat testis.
- Frank French (6 December 2012). Hormonal Regulation of Spermatogenesis. Springer Science & Business Media. pp. 159–. ISBN 978-1-4613-4440-7.
Consequently, we infused Medrogestone into the perfused testis since Medrogestone is a potent inhibitor of the Δ5-3-ketosteroid isomerase enzyme (52) and should therefore inhibit testosterone biosynthesis.
- Canadian Federation of Biological Societies. Proceedings of the Annual Meeting.
With a rat testicular enzyme preparation, Medrogestone (lxlO'^M) inhibited the conversion of pregnenolone to progesterone (92%) and 17- hydroxyprogesterone (59%); and 17-hydroxy- pregnenolone to 17- hydroxyprogesterone (83%). With a ...
- Influences of Hormones in Tumor Development. CRC Press. 1979. ISBN 978-0-8493-5352-9.
Pharmacologic doses of medrogestone can inhibit 5a reduction of testosterone by microsomal preparations from the skin and the prostate.
- Benign Prostatic Hypertrophy. Springer Science & Business Media. 6 December 2012. pp. 266–. ISBN 978-1-4612-5476-8.
medrogestone has marked antiandrogenic activity as well, although not as much as flutamide or cyproterone acetate.
- Deghenghi R, Revesz C, Gaudry R (May 1963). "New Synthesis and Structure Activity Relationship in the 17-Alkylated Progesterone Series". Journal of Medicinal Chemistry. 6 (3): 301–4. doi:10.1021/jm00339a019. PMID 14185989.
- "Drugs@FDA: FDA Approved Drug Products". United States Food and Drug Administration. Retrieved 16 November 2016.