Laron syndrome
Laron syndrome, or Laron-type dwarfism, is an autosomal recessive disorder characterized by an insensitivity to growth hormone (GH), usually caused by a mutant growth hormone receptor. It causes a short stature and an increased sensitivity to insulin which means that diabetes mellitus type 2 is less likely to develop,[1] and possibly cancer as well. It can be treated with injections of recombinant IGF-1.
| Laron syndrome | |
|---|---|
| Other names | Laron-type dwarfism |
 | |
| Growth hormone | |
| Specialty | Endocrinology |
Presentation
The principal feature of Laron syndrome is abnormally short stature (dwarfism). Physical symptoms include: prominent forehead, depressed nasal bridge, underdevelopment of mandible, truncal obesity,[2] and micropenis in males. The breasts of females reach normal size, and in some are large in relation to body size.[3] It has been suggested that hyperprolactinemia may contribute to the enlarged breast size.[4] Seizures are frequently seen secondary to hypoglycemia. Some genetic variations decrease intellectual capacity.[5] Laron syndrome patients also do not develop acne, except temporarily during treatment with IGF-1 (if performed).[4]
In 2011, it was reported that people with this syndrome in the Ecuadorian villages are resistant to cancer and diabetes and are somewhat protected against aging.[6][7][8] This is consistent with findings in mice with a defective growth hormone receptor gene.[9]
Pathophysiology
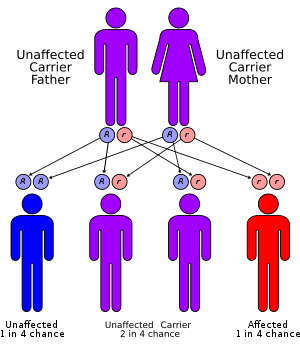
Molecular genetic investigations have shown that this disorder is mainly associated with mutations in the gene for the GH receptor. These can result in defective hormone binding to the ectodomain or reduced efficiency of dimerization of the receptor after hormone occupancy. There are exceptionally low levels of insulin-like growth factor (IGF-1) and its principal carrier protein, insulin-like growth factor binding protein 3.
A related condition involving postreceptor insensitivity to growth hormone has been associated with STAT5B.[10]
Diagnosis
Laron syndrome can be diagnosed through biochemical analyses: basal GH levels are abnormally increased, while IGF-1 is low. When a GHRH test is performed, IGF-1 fails to increase. GHBP (growth hormone binding protein) levels are low in cases with mutations in the extracellular domain of the GH receptor and normal in cases with mutations in the intracellular domain. Genetic tests should be performed to make a precise etiological diagnosis.
Treatment
Administration of GH has no effect on IGF-1 production, therefore treatment is mainly by biosynthetic IGF-1. IGF-1 must be taken before puberty to be effective.[9]
The drug product Increlex (mecasermin), developed by the company Tercica, purchased by Ipsen, was approved by the US Food and Drug Administration in August 2005 for replacing IGF-1 in patients who are deficient.[11]
IPLEX (Mecasermin rinfabate) is composed of recombinant human IGF-1 (rhIGF-1) and its binding protein IGFBP-3. It was approved by the U.S. Food and Drug Administration (FDA) in 2005 for treatment of primary IGF-1 deficiency or GH gene deletion.[12][13] Side effects from IPLEX are hypoglycemia. IPLEX's manufacturing company, Insmed, after selling its protein production facility, can no longer develop proteins, thus can no longer manufacture IPLEX as of a statement released in July 2009.[14]
Prognosis
People with Laron syndrome have strikingly low rates of cancer and diabetes, although they appear to be at increased risk of accidental death due to their stature.[9][7]
Incidence
The majority of reported cases of Laron syndrome have been in people with Semitic origins, almost all of them being Jews or assimilated descendants of Jews.
Numerous Laron syndrome patients are found in Israel among the country's diverse Jewish population composed of Jews from around the world, as well as patients outside Israel originally from communities of the Jewish diaspora, such as Egypt and Iraq. There is also a disproportionate number of sufferers found in remote villages in Ecuador who are descended from colonial-era Jewish-origin New Christian conversos (Sephardi Jews who themselves, or whose forebears, had been compelled to convert to Catholicism back in Spain) who had covertly migrated to Ecuador during the Spanish Conquest despite the Spanish Crown's prohibition of their emigration to its colonies and territories as a result of the Inquisition.[6][9]
Other patients include people of other Semitic non-Jewish origins, including from Saudi Arabia.
Eponym
It is named after Zvi Laron, the Israeli researcher who, with A. Pertzelan and S. Mannheimer, first reported the condition in 1966,[15][16] based upon observations which began in 1958.[17]
Resistance to GH was first reported by Laron in 1966. Since then, severe resistance to GH, characterized by grossly impaired growth despite normal levels of GH in serum, has been termed Laron syndrome.
Homo floresiensis
Recent publications have proposed that Homo floresiensis represented a population with widespread Laron syndrome.[18][19] This is only one of several competing hypotheses, and has received criticism as insufficient to explain the "range features observed in H. floresiensis".[20]
See also
References
- Bartke, Andrzej; Sun, Liou Y.; Longo, Valter (April 2013). "Somatotropic Signaling: Trade-Offs Between Growth, Reproductive Development, and Longevity". Physiological Reviews. 93 (2): 571–598. doi:10.1152/physrev.00006.2012. PMC 3768106. PMID 23589828.
- Laron Z, Ginsberg S, Lilos P, Arbiv M, Vaisman N (2006). "Body composition in untreated adult patients with Laron syndrome (primary GH insensitivity)". Clin. Endocrinol. 65 (1): 114–7. doi:10.1111/j.1365-2265.2006.02558.x. PMID 16817829.
- Laron, Zvi (2004). "Laron Syndrome (Primary Growth Hormone Resistance or Insensitivity): The Personal Experience 1958–2003". The Journal of Clinical Endocrinology & Metabolism. 89 (3): 1031–1044. doi:10.1210/jc.2003-031033. ISSN 0021-972X. PMID 15001582.
- Zvi Laron; J. Kopchick (25 November 2010). Laron Syndrome - From Man to Mouse: Lessons from Clinical and Experimental Experience. Springer Science & Business Media. pp. 253, 255. ISBN 978-3-642-11183-9.
- Shevah O, Kornreich L, Galatzer A, Laron Z (2005). "The intellectual capacity of patients with Laron syndrome (LS) differs with various molecular defects of the growth hormone receptor gene. Correlation with CNS abnormalities". Horm. Metab. Res. 37 (12): 757–60. doi:10.1055/s-2005-921097. PMID 16372230.
- Guevara-Aguirre, J; Balasubramanian, P; Guevara-Aguirre, M; Wei, M; Madia, F; Cheng, CW; Hwang, D; Martin-Montalvo, A; et al. (2011). "Growth Hormone Receptor Deficiency Is Associated with a Major Reduction in Pro-Aging Signaling, Cancer, and Diabetes in Humans". Science Translational Medicine. 3 (70): 70ra13. doi:10.1126/scitranslmed.3001845. PMC 3357623. PMID 21325617.
- Bai, Nina. "Defective Growth Gene in Rare Dwarfism Disorder Stunts Cancer and Diabetes". Scientific American. Retrieved 17 February 2011.
- Winerman, Lea. "Study: Dwarfism Gene May Offer Protection From Cancer, Diabetes". PBS. Retrieved 17 February 2011.
- Wade, Nicholas (17 February 2011). "Ecuadorean Villagers May Hold Secret to Longevity". The New York Times. ISSN 0362-4331. Retrieved 17 February 2011.
- Hwa V, Camacho-Hübner C, Little BM, et al. (2007). "Growth hormone insensitivity and severe short stature in siblings: a novel mutation at the exon 13-intron 13 junction of the STAT5b gene". Horm. Res. 68 (5): 218–24. doi:10.1159/000101334. PMID 17389811.
- "Increlex (mecasermin)". Centerwatch.com. Retrieved 21 November 2014.
- Kemp, S.F. "Mecasermin rinfabate". Thomson Reuters. Retrieved 5 March 2011.
- Meyer, Robert. "Approval letter (Mecasermin rinfabate)" (PDF). FDA. Retrieved 5 March 2011.
- "Insmed Provides Update on Supply of IPLEX(TM)". Retrieved 25 August 2017.
- synd/2825 at Who Named It?
- Laron Z, Pertzelan A, Mannheimer S (1966). "Genetic pituitary dwarfism with high serum concentration of growth hormone—a new inborn error of metabolism?". Isr. J. Med. Sci. 2 (2): 152–5. PMID 5916640.
- Laron Z (2004). "Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958–2003". J. Clin. Endocrinol. Metab. 89 (3): 1031–44. doi:10.1210/jc.2003-031033. PMID 15001582.
- Hershkovitz I, Kornreich L, Laron Z (2007). "Comparative skeletal features between Homo floresiensis and patients with primary growth hormone insensitivity (Laron syndrome)". Am. J. Phys. Anthropol. 134 (2): 198–208. doi:10.1002/ajpa.20655. PMID 17596857.
- Culotta E (2007). "Paleoanthropology. The fellowship of the hobbit". Science. 317 (5839): 740–742. doi:10.1126/science.317.5839.740. PMID 17690271.
- Aiello, Leslie C. (2010). "Five years ofHomo floresiensis". American Journal of Physical Anthropology. 142 (2): 167–79. doi:10.1002/ajpa.21255. ISSN 0002-9483. PMID 20229502.
External links
| Classification | |
|---|---|
| External resources |
- Laron+syndrome at the US National Library of Medicine Medical Subject Headings (MeSH)