Interrupted aortic arch
Interrupted aortic arch is a very rare heart defect (affecting 3 per million live births)[1] in which the aorta is not completely developed. There is a gap between the ascending and descending thoracic aorta. In a sense it is the complete form of a coarctation of the aorta. Almost all patients also have other cardiac anomalies, including a ventricular septal defect (VSD), aorto-pulmonary window, and truncus arteriosus. There are three types of interrupted aortic arch, with type B being the most common. Interrupted aortic arch (especially Type B) is often associated with DiGeorge syndrome.[2]
| Interrupted aortic arch | |
|---|---|
 | |
| Specialty | Cardiology |
Causes
It is thought that an interrupted aortic arch occurs through excessive apoptosis in the developing, embryonic aorta.[3]
Diagnosis
It can be diagnosed with a standard echocardiogram.[2] An echocardiogram can also aid in classifying the type of defect.[2] The diagnosis can also be made prior to birth via ultrasound.[3] Patients will have a loss of appetite, appear tired and weak, and exhibit rapid breathing and a rapid heart rate.[4] If the condition progresses, the infant may turn pale, feel cold in the lower half of the body, and have a weak pulse due to insufficient blood flow.[4] The pattern of pulse abnormalities is dependent upon the classification; e.g., for type B interrupted aortic arch, the right brachial pulse will be palpable and the left brachial and femoral pulses will be impalpable due to closure of the ductus arteriosus.[3] Rarely, an interrupted aortic arch can be associated with an intracranial aneurysm.[5] Signs of ischemia due to interrupted aortic arch can be separated by the organ system involved:[3]
- Liver injury: elevated serum glutamic oxaloacetic transaminase (SGOT) (also known as aspartate transaminase, AST) and lactic acid dehydrogenase (LDH)
- Kidney injury: elevated serum creatinine
- Intestinal injury: signs of necrotizing enterocolitis, such as bloody stools
CHARGE syndrome, a specific, rare pattern of genetic abnormalities, commonly features conotruncal and aortic arch heart defects, which can include an interrupted aortic arch.[6]
Classification
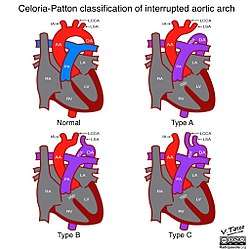
There are three primary classifications for an interrupted aortic arch, on the basis of the specific, anatomic anomaly.[4] They are:
- Type A: The aortic arch is interrupted after the left subclavian artery.[4]
- Type B: The aortic arch is interrupted between the left common carotid artery and the left subclavian artery. This is the most common form of the condition, and is the classification most often associated with DiGeorge syndrome.[4]
- Type C: The aortic arch is interrupted between the innominate artery and the left common carotid artery. This is the least common form of the condition.[4]
Each class can be divided into two subgroups, based upon whether the right subclavian artery originated in a normal, anatomical position (subgroup 1) or if it originated distal to the left subclavian artery and continues behind the esophagus (subgroup 2).[7] However, these subgroups do not affect how the disease is diagnosed or treated.[7]
Treatment
If the diagnosis is made prenatally, prostaglandin E1 (PGE1) is started after birth to avoid closure of the ductus arteriosus.[3] Prostaglandin therapy is performed via a continuous infusion, due to how quickly prostaglandins are metabolized in the body.[8] However, the diagnosis may go undetected, delaying treatment until closure of the ductus arteriosus produces symptoms.[3]
Curative treatment consists of open heart surgery soon after birth, preferably within the first week after birth while there is a patent ductus arteriosus. Awaiting surgery, prostaglandin can be administered to keep the ductus arteriosus open, thereby allowing blood flow to the lower body. After successful treatment, the patient is monitored for the rest of their life by a specialist to ensure that problems do not occur.[4]
Prognosis
Failure to treat the condition yields a mortality rate of 90% at a median age of 4 days.[1] Death occurs due to increased blood flow from the left side of the heart (oxygenated blood) to the right side (deoxygenated blood), inducing heart failure; pulmonary edema; and eventual closing of the ductus arteriosus.[7] For an infant with an interrupted aortic arch, a patent (open) ductus arteriosus allows for blood to bypass the "interruption," without which blood will be unable to reach the lower half of the body.[9] As a result, the kidneys fail and the blood becomes acidic, resulting in death.[7]
With modern surgical techniques, 81% of children with an interrupted aortic arch survive to be 15 years-old.[5] The fate of survivors in the long-term is still unclear.[1]
Surgical complications
The most common, early complication of surgery is bleeding, the risk of which can be increased by prematurity, prolonged acidosis prior to surgery, and excessive tension on the anastamosis due to inadequate mobilization of the ascending and descending aorta.[3] Other early complications include damage to the left recurrent laryngeal nerve and the phrenic nerve.[3] Late complications include obstruction of the graft and obstruction of the left main bronchus (which passes underneath the aortic arch).[3]
Epidemiology
The incidence of an interrupted aortic arch is extremely rare, occurring between three[1] and twenty times per 1,000,000 births.[10] In the context of other congenital cardiac abnormalities, interrupted aortic arch represents about 1.5% of cases.[3]
History
The condition was first identified by Dr. Raphael Steidele, Professor of Obstetrics at the University of Vienna, in 1778.[11] In the case Steidele described, the infant had a type A interrupted aortic arch, and survived only for "a few hours."[11] In homage to the discoverer, the terminology of "Steidele's complex" has been used to describe an interrupted aortic arch.[11] The first type B interrupted aortic arch was reported by Seidel in 1818, and the first type C was reported by Weisman and Kesten in 1948.[7] The classification system (Types A, B, and C) were defined by Celoria and Patton in 1959.[1]
The first successful repair of a Type A interrupted aortic arch was reported in 1961, in which the left subclavian artery was grafted into the descending thoracic aorta in a 14-year-old male patient.[7] The first successful repair of a Type A interrupted aortic arch in an infant was in a 12-day-old infant in 1969, in which the left subclavian artery was connected to the descending thoracic aorta, the patent ductus arteriosus was closed, and the main pulmonary artery was banded.[7]
The first successful repair of a Type B interrupted aortic arch was in 1954, in which the 16 year-old, female patient's own aorta was grafted from the arch to the descending thoracic aorta and the left subclavian artery was ligated.[7] The first successful repair on a type B interrupted aortic arch in an infant was in 1973, in which a vein was used to connect the ascending aorta and the descending aorta.[7]
The first successful repair of a Type C interrupted aortic arch was in 1964, in which the 16-year-old female patient's ascending aorta was grafted to the descending thoracic aorta.[7] As of 1984, there had been no successful repairs on infants under one year old.[7]
The use of PGE1 dramatically improved the mortality rate after its introduction in 1976.[3]
Research directions
While PGE1 is the standard of care for maintaining the ductus arteriosus, there is insufficient data on the proper dose, duration of therapy, safety, and long-term consequences of PGE1 on infants with ductal-dependent congenital heart defects (like interrupted aortic arch).[12]
References
- Messner, Greg; Reul, George J.; Flamm, Scott D.; Gregoric, Igor D.; Opfermann, Ulrich Tim (2002). "Interrupted aortic arch in an adult single-stage extra-anatomic repair". Texas Heart Institute Journal / From the Texas Heart Institute of St. Luke's Episcopal Hospital, Texas Children's Hospital. 29 (2): 118–21. PMC 116738. PMID 12075868.
- Chin, Alvin J (Oct 2, 2007). "Interrupted Aortic Arch". eMedicine. Retrieved May 27, 2009.
- Jonas, Richard (2013). "15". In Mavroudis, Constantine; Backer, Carl; Idriss, Richid (eds.). Pediatric Cardiac Surgery (4th ed.). Chichester: Wiley-Blackwell. pp. 283–294. ISBN 9781118320754.
- "Interrupted Aortic Arch | Types, Treatment & Repair". www.cincinnatichildrens.org. Cincinnati Children's Hospital Medical Center. July 2015. Retrieved 28 April 2018.
- Börcek, Alp Özgün; Egemen, Emrah; Güngör, Günhan; Baykaner, Mustafa Kemali (6 November 2012). "Intracranial aneurysm in childhood and interrupted aortic arch". Child's Nervous System. 29 (1): 11–15. doi:10.1007/s00381-012-1959-6. PMID 23129447.
- Blake, Kim D.; Davenport, Sandra L. H.; Hall, Bryan D.; Hefner, Margaret A.; Pagon, Roberta A.; Williams, Marc S.; Lin, Angela E.; Graham, John M. (2 July 2016). "CHARGE Association: An Update and Review for the Primary Pediatrician". Clinical Pediatrics. 37 (3): 159–173. doi:10.1177/000992289803700302. PMID 9545604.
- Reardon, MJ; Hallman, GL; Cooley, DA (September 1984). "Interrupted aortic arch: brief review and summary of an eighteen-year experience". Texas Heart Institute Journal. 11 (3): 250–9. PMC 341721. PMID 15227058.
- Sharma, Mukti; Sasikumar, M; Karloopia, SD; Shahi, BN (April 2001). "Prostaglandins in congenital heart disease". Medical Journal Armed Forces India. 57 (2): 134–138. doi:10.1016/S0377-1237(01)80134-9. PMC 4925861. PMID 27407318.
- "Interrupted Aortic Arch". www.mottchildren.org. University of Michigan. Retrieved 28 April 2018.
- "Interrupted Aortic Arch". Cleveland Clinic. Cleveland Clinic. Retrieved 28 April 2018.
- Lie, J.T. (May 1967). "The malformation complex of the absence of the arch of the aorta-Steidele's complex". American Heart Journal. 73 (5): 615–625. doi:10.1016/0002-8703(67)90171-8.
- Akkinapally, Smita; Hundalani, Shilpa G; Kulkarni, Madhulika; Fernandes, Caraciolo J; Cabrera, Antonio G; Shivanna, Binoy; Pammi, Mohan (27 February 2018). "Prostaglandin E1 for maintaining ductal patency in neonates with ductal-dependent cardiac lesions". Cochrane Database of Systematic Reviews. 2 (2): CD011417. doi:10.1002/14651858.CD011417.pub2. PMC 6491149. PMID 29486048.
Further reading
- Collins-Nakai, RL; Dick, M; Parisi-Buckley, L; Fyler, DC; Castaneda, AR (1976). "Interrupted aortic arch in infancy". The Journal of Pediatrics. 88 (6): 959–62. doi:10.1016/S0022-3476(76)81049-9. PMID 1271195.
External links
| Classification | |
|---|---|
| External resources |