IPEX syndrome
Immunodysregulation polyendocrinopathy enteropathy X-linked (or IPEX) syndrome is a rare disease linked to the dysfunction of the transcription factor FOXP3, widely considered to be the master regulator of the regulatory T cell lineage.[4][5] It leads to the dysfunction of regulatory T-cells and the subsequent autoimmunity.[6] The disorder is one of the autoimmune polyendocrine syndromes and manifests with autoimmune enteropathy, psoriasiform or eczematous dermatitis, nail dystrophy, autoimmune endocrinopathies, and autoimmune skin conditions such as alopecia universalis and bullous pemphigoid.[6][7] Management for IPEX has seen limited success in treating the syndrome by bone marrow transplantation.[8]
| IPEX syndrome | |
|---|---|
| Other names | Autoimmune enteropathy type 1[1] |
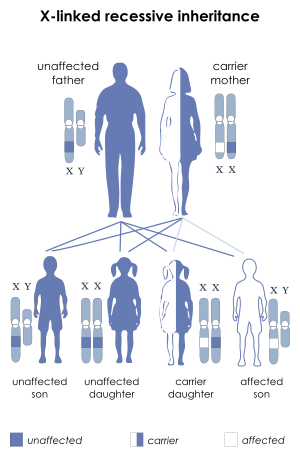 | |
| IPEX syndrome is inherited via X-linked recessive | |
| Specialty | Immunology |
| Causes | FOXP3 gene mutation[1] |
| Diagnostic method | Family history, Genetic test[1] |
| Treatment | TPN(nutritional purpose), Cyclosporin A and FK506, Bone marrow transplant[2][3] |

Genetics
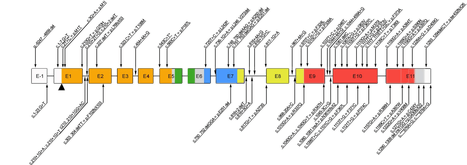
IPEX syndrome is inherited in males via an x-linked recessive manner, as the FOXP3 gene, whose cytogenetic location is Xp11.23, is involved in the mechanism of this condition.[4][5]
Mutation of FOXP3 leading to expression of malfunctioning protein is often localised in DNA-binding domain called the forkhead domain. The truncated protein can not bind to its binding-spot on the DNA and thus its function concerning T regulatory lymphocytes development and functioning is impaired. The absence or dysfunction of regulatory T cells is the cause of autoimmune symptoms.[9]
Data from 2018 describes over 70 mutations in FOXP3 gene leading to IPEX syndrome. Nonetheless, this number is still changing with new cases and discoveries coming.[10] For example in 2010 there were only 20 mutations of FOXP3 known in the literature.[9]
Mechanism
This autoimmunity called IPEX is an attack from the body's own immune system against the body's own tissues and organs.[3] Early age onset of this disease in males causes severe enlargement of the secondary lymphoid organs, and insulin dependent diabetes
This condition indicates the loss of CD4+CD25+ T regulatory cells, and express the transcription factor Foxp3. Foxp3 decrease is a consequence of unchecked T cell activation, which is secondary to loss of regulatory T cells.[11]
Diagnosis
The diagnosis of immunodysregulation polyendocrinopathy enteropathy X-linked syndrome is consistent with the following criteria:[1]
- Clinical examination
- Family history
- Laboratory findings
- Genetic testing
Treatment
In terms of treatment the following are done to manage the IPEX syndrome in those affected individuals (corticosteroids are the first treatment that is used):[3][2]
- TPN (nutritional purpose)
- Cyclosporin A and FK506
- Sirolimus (should FK506 prove non-effective)
- Granulocyte colony stimulating factor
- Bone marrow transplant
- Rituximab
Research
There is as well a special mouse model simulating the development and progression of the IPEX syndrome. The model mice are called "scurfy mice" and they have had 2 base pairs inserted within the Foxp3 gene. Consequently, this leads to a frameshift and the expressed protein is truncated, causing the same effects as FOXP3 mutation in humans. The mice suffer from enlarged spleen and lymph nodes, redness in eyes and skin abnormalities. The mice as well suffer from immunity problems and after approximately 3 weeks they die.[10]
References
- RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Immune dysregulation polyendocrinopathy enteropathy X linked syndrome". www.orpha.net. Retrieved 2017-04-18.
- Eisenbarth, George S. (2010-12-13). Immunoendocrinology: Scientific and Clinical Aspects. Springer Science & Business Media. pp. 129–138. ISBN 9781603274784.
- Hannibal, Mark C.; Torgerson, Troy (1993-01-01). "IPEX Syndrome". In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Ledbetter, Nikki; Mefford, Heather C. (eds.). GeneReviews. Seattle (WA): University of Washington, Seattle. PMID 20301297.update 2011
- Reference, Genetics Home. "IPEX syndrome". Genetics Home Reference. Retrieved 2017-04-16.
- Reference, Genetics Home. "FOXP3 gene". Genetics Home Reference. Retrieved 2017-04-16.
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. p. 72. ISBN 978-1-4160-2999-1.
- "Immunodysregulation, polyendocrinopathy and enteropathy X-linked | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2017-04-16.
- Wildin RS, Smyk-Pearson S, Filipovich AH (August 2002). "Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome". J Med Genet. 39 (8): 537–45. doi:10.1136/jmg.39.8.537. PMC 1735203. PMID 12161590.
- Michels, Aaron W.; Gottlieb, Peter A. (2010). "Autoimmune polyglandular syndromes". Nature Reviews Endocrinology. 6 (5): 270–277. doi:10.1038/nrendo.2010.40. ISSN 1759-5029. PMID 20309000.
- Bacchetta, Rosa; Barzaghi, Federica; Roncarolo, Maria-Grazia (2018). "From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation: IPEX syndrome and FOXP3". Annals of the New York Academy of Sciences. 1417 (1): 5–22. doi:10.1111/nyas.13011. PMID 26918796.
- Verbsky, James W.; Chatila, Talal A. (2017-04-18). "Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-linked (IPEX) and IPEX-Related Disorders: an Evolving Web of Heritable Autoimmune Diseases". Current Opinion in Pediatrics. 25 (6): 708–714. doi:10.1097/MOP.0000000000000029. ISSN 1040-8703. PMC 4047515. PMID 24240290.
Further reading
- Bacchetta, Rosa; Barzaghi, Federica; Roncarolo, Maria-Grazia (25 February 2016). "From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation". Annals of the New York Academy of Sciences. 1417 (1): 5–22. doi:10.1111/nyas.13011. ISSN 1749-6632. PMID 26918796.
- Barzaghi, Federica; Passerini, Laura; Bacchetta, Rosa (1 January 2012). "Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome: A Paradigm of Immunodeficiency with Autoimmunity". Frontiers in Immunology. 3: 211. doi:10.3389/fimmu.2012.00211. ISSN 1664-3224. PMC 3459184. PMID 23060872.
- Elzouki, A. Y.; Harfi, H. A.; Nazer, H.; Stapleton, F. B.; Oh, William; Whitley, R. J. (2012-01-10). Textbook of Clinical Pediatrics. Springer Science & Business Media. ISBN 9783642022029.
External links
| Classification | |
|---|---|
| External resources |
|