Hunter syndrome
Hunter syndrome, or mucopolysaccharidosis type II (MPS II), is a rare genetic disorder in which large sugar molecules called glycosaminoglycans (AKA GAGs, or mucopolysaccharides) build up in body tissues. It is a form of lysosomal storage disease. Hunter Syndrome is caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase (I2S).[2][3] The lack of this enzyme causes heparan sulfate and dermatan sulfate to accumulate in all body tissues.[4] Hunter syndrome is the only MPS syndrome to exhibit X-linked recessive inheritance.[4]
| Hunter syndrome | |
|---|---|
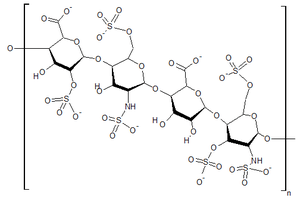 | |
| Structure of heparan sulfate, one of the GAGs that builds up in the tissues of people with Hunter syndrome | |
| Specialty | Endocrinology |
| Symptoms | Skeletal abnormalities, hearing loss, retinal degeneration, enlarged liver and spleen |
| Complications | Upper airway disease; cardiovascular failure |
| Causes | Defiency of the enzyme iduronate-2-sulfatase |
| Differential diagnosis | Mucopolysaccharidosis type I; other mucopolysaccharidoses |
| Prognosis | In severe cases, death usually occurs by age 15. In attenuated cases, patients may survive into their 50s |
| Frequency | 1 in 100,000 to 150,000 male births[1] |
The symptoms of Hunter syndrome are comparable to those of MPS I. Hunter syndrome causes abnormalities in many organs, including the skeleton, heart, and respiratory system. In severe cases, this leads to death during the teenage years. Unlike MPS I, corneal clouding is not associated with this disease.[1]
Signs and symptoms
Hunter syndrome may present with a wide variety of phenotypes. Hunter syndrome has traditionally been categorized as either "mild" or "severe" depending on the presence of central nervous system symptoms. However, this is an oversimplification. Patients with "attenuated" or "mild" forms of the disease may still suffer from significant health issues. For severely affected patients, the clinical course is relatively predictable; patients will normally die at an early age. For those with milder forms of the disease, there is a wider variety of outcomes. Many live into their 20s and 30s, but some may have near-normal life expectancies and may even have children. Cardiac and respiratory abnormalities are the usual cause of death for patients with milder forms of the disease.[2]
The symptoms of Hunter syndrome (MPS II) are generally not apparent at birth. Often, the first symptoms may include abdominal hernias, ear infections, runny noses, and colds. As the buildup of GAGs continues throughout the cells of the body, signs of Hunter syndrome become more visible. Physical appearances of many children with Hunter syndrome include a distinctive coarseness in their facial features, including a prominent forehead, a nose with a flattened bridge, and an enlarged tongue. They may also have a large head, as well as an enlarged abdomen. For severe cases of MPS II, a diagnosis is often made between the ages of 18 and 36 months. In milder cases, patients present similarly to children with Hurler–Scheie syndrome, and a diagnosis is usually made between the ages of 4 and 8 years.[2]
The continued storage of GAGs leads to abnormalities in multiple organ systems. After 18 months, children with severe MPS II may suffer from developmental decline and progressive loss of skills.[1] The thickening of the heart valves and walls of the heart can result in progressive decline in cardiac function. The walls of the airway may become thickened, as well, leading to obstructive airway disease. As the liver and spleen grow larger with time, the abdomen may become distended, making hernias more noticeable. All major joints may be affected by Hunter syndrome, leading to joint stiffness and limited motion. Progressive involvement of the finger and thumb joints results in decreased ability to pick up small objects. The effects on other joints, such as hips and knees, can make walking normally increasingly difficult. If carpal tunnel syndrome develops, a further decrease in hand function can occur. The bones themselves may be affected, resulting in short stature. In addition, pebbly, ivory-colored skin lesions may be found on the upper arms, legs, and upper back of some people with Hunter syndrome. These skin lesions are considered pathognomic for the disease. Finally, the storage of GAGs in the brain can lead to delayed development with subsequent mental retardation and progressive loss of function.
The age at onset of symptoms and the presence/absence of behavioral disturbances are predictive factors of ultimate disease severity in very young patients. Behavioral disturbances can often mimic combinations of symptoms of attention deficit hyperactivity disorder, autism, obsessive compulsive disorder, and/or sensory processing disorder, although the existence and level of symptoms differ in each affected child. They often also include a lack of an appropriate sense of danger, and aggression. The behavioral symptoms of Hunter syndrome generally precede neurodegeneration and often increase in severity until the mental handicaps become more pronounced.[5] By the time of death, most children with severe MPS II have severe mental disabilities and are completely dependent on their caretakers.[2]
Genetics
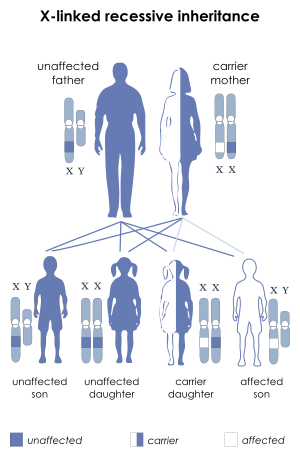
Since Hunter syndrome is an X-linked recessive disorder, it preferentially affects male patients. The IDS gene is located on the X chromosome. The IDS gene encodes for an enzyme called iduronate-2-sulfatase (I2S). A lack of this enzyme leads to a buildup of GAGs, which cause the symptoms of Hunter syndrome.[6] Females generally have two X chromosomes, whereas males generally have one X chromosome that they inherit from their mother and one Y chromosome that they inherit from their father.
If a female inherits one copy of the mutant allele for MPS II, she will usually have a normal copy of the IDS gene which can compensate for the mutant allele. This is known as being a genetic carrier. However, a male who inherits a defective X chromosome usually does not have another X chromosome to compensate for the mutant gene. Thus, a female would need to inherit two mutant genes in order to develop Hunter Syndrome, while a male patient only needs to inherit one mutant gene. However, it is possible for a female carrier to be affected due to X-inactivation, which is a random process.
Pathophysiology
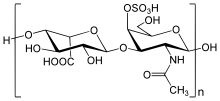
The human body depends on a vast array of biochemical reactions to support critical functions. One of these functions is the breakdown of large biomolecules. The failure of this process is the underlying problem in Hunter syndrome and related storage disorders.
The biochemistry of Hunter syndrome is related to a problem in a part of the connective tissue known as the extracellular matrix. This matrix is made up of a variety of sugars and proteins. It helps to form the architectural framework of the body. The matrix surrounds the cells of the body in an organized meshwork and functions as the glue that holds the cells of the body together. One of the parts of the extracellular matrix is a molecule called a proteoglycan. Like many components of the body, proteoglycans need to be broken down and replaced. When the body breaks down proteoglycans, one of the resulting products is mucopolysaccharides (GAGs).
In Hunter syndrome, the problem concerns the breakdown of two GAGs: dermatan sulfate and heparan sulfate. The first step in the breakdown of dermatan sulfate and heparan sulfate requires the lysosomal enzyme iduronate-2-sulfatase, or I2S. In people with Hunter syndrome, this enzyme is either partially or completely inactive. As a result, GAGs build up in cells throughout the body, particularly in tissues that contain large amounts of dermatan sulfate and heparan sulfate. The rate of GAGs buildup is not the same for all people with Hunter syndrome, resulting in a wide spectrum of medical problems.
Diagnosis
The first laboratory screening test for an MPS disorder is a urine test for GAGs. Abnormal values indicate that an MPS disorder is likely. The urine test can occasionally be normal even if the child actually has an MPS disorder. A definitive diagnosis of Hunter syndrome is made by measuring I2S activity in serum, white blood cells, or fibroblasts from skin biopsy. In some people with Hunter syndrome, analysis of the I2S gene can determine clinical severity.
Prenatal diagnosis is routinely available by measuring I2S enzymatic activity in amniotic fluid or in chorionic villus tissue. If a specific mutation is known to run in the family, prenatal molecular genetic testing can be performed. DNA sequencing can reveal if someone is a carrier for the disease.[2]
Treatment
Because of the wide variety of phenotypes, the treatment for this disorder is specifically determined for each patient. Until recently, there was no effective therapy for MPS II. Because of this, palliative care was used. However, recent advances have led to medications which can improve survival and well-being in people with MPS II.
Enzyme replacement therapy
Idursulfase, a purified form of the missing lysosomal enzyme, underwent clinical trial in 2006[6] and was subsequently approved by the United States Food and Drug Administration as an enzyme replacement treatment for Hunter syndrome. Idursulfase beta, another enzyme replacement treatment, was approved in Korea by the Ministry of Food and Drug Safety.
Recent advances in enzyme replacement therapy (ERT) with idursulfase have been proven to improve many signs and symptoms of MPS II, especially if started early in the disease. After administration, it can be transported into cells in order to break down GAGs. However, as the medication cannot cross the blood–brain barrier, it is not expected to lead to cognitive improvement in patients with severe central nervous system symptoms. Even with ERT, treatment of various organ problems from a wide variety of medical specialists is necessary.[2]
Bone marrow and stem cell transplantation
Bone marrow transplantation and hematopoietic stem cell transplantation (HSCT) have been used as treatments in some studies.[7][8] While transplantation has provided benefits for many organ systems, it has not been shown to improve the neurological symptoms of the disease. Although HSCT has shown promise in the treatment of other MPS disorders, its results have been unsatisfactory so far in the treatment of MPS II. ERT has been shown to lead to better outcomes in MPS II patients.[2]
Gene editing therapy
In February 2019, medical scientists working with Sangamo Therapeutics, headquartered in Richmond, California, announced the first ever "in body" human gene editing therapy to permanently alter DNA - in a patient with Hunter Syndrome.[9] Clinical trials by Sangamo involving gene editing using Zinc Finger Nuclease (ZFN) are ongoing.[10]
Prognosis
Earlier onset of symptoms is linked to a worse prognosis. For children who exhibit symptoms between the ages of 2 to 4, death usually occurs by the age of 15 to 20 years. The cause of death is usually due to neurological complications, obstructive airway disease, and cardiac failure. If patients have minimal neurologic involvement, they may survive into their 50s or beyond.[1][6]
Epidemiology
There are estimated to be approximately 2,000 people afflicted with Hunter syndrome worldwide, 500 of whom live in the United States.[11]
A study in the United Kingdom indicated an incidence among males of approximately 1 in 130,000 male live births.[12]
History
The syndrome is named after physician Charles A. Hunter (1873–1955), who first described it in 1917.[13][14]
Research
Beginning in 2010, a phase I/II clinical trial evaluated intrathecal injections of a more concentrated dose of idursulfase than the intravenous formulation used in enzyme replacement therapy infusions, in hopes of preventing the cognitive decline associated with the severe form of the condition.[15] Results were reported in October 2013.[16] A phase II/III clinical trial began in 2014.[17]
In 2017, a 44-year-old[18] patient with Hunter syndrome was treated with gene therapy in an attempt to prevent further damage by the disease. This is the first case of gene therapy being used in vivo in humans.[19] The study was extended to six patients in 2018.[20]
Society
On 24 July 2004 Andrew Wragg, 38, of Worthing, West Sussex, England, suffocated his 10-year-old son Jacob with a pillow, because of the boy's disabilities related to Hunter syndrome. A military security specialist, Wragg also claimed that he was under stress after returning from the war in Iraq. He denied murdering Jacob, but pleaded guilty to manslaughter by reason of diminished capacity. Mrs. Justice Anne Rafferty, called the case "exceptional", gave Wragg a two-year prison sentence for manslaughter, then suspended his sentence for two years. Rafferty said there was "nothing to be gained" from sending Wragg to prison for the crime.[21][22][23] On 13 December 2005 Andrew Wragg walked out of Lewes Crown Court a free man after a jury determined that he did not murder his 10-year-old son.
See also
- Hurler syndrome (MPS I)
- Sanfilippo syndrome (MPS III)
- Morquio syndrome (MPS IV)
- Prenatal testing
- Genetic counseling
References
- "Mucopolysaccharidoses Fact Sheet". National Institute of Neurological Disorders and Stroke. 15 Nov 2017. Retrieved 11 May 2018.
- Wraith JE, Scarpa M, Beck M, et al. (March 2008). "Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy". Eur. J. Pediatr. 167 (3): 267–77. doi:10.1007/s00431-007-0635-4. PMC 2234442. PMID 18038146.
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. p. 544. ISBN 978-0-7216-2921-6.
- Le, Tao; Bhushan, Vikas; Hofmann, Jeffrey (2012). First Aid for the USMLE Step 1. McGraw-Hill. p. 117.
- Schwartz, Ida VD (2007). "A clinical study of 77 patients with mucopolysaccharidosis type II". Acta Paediatrica. 96 (455): 63–70. doi:10.1111/j.1651-2227.2007.00212.x. PMID 17391446.
- Muenzer, J; Wraith, JE; Beck, M; Giugliani, R; Harmatz, P; Eng, CM; Vellodi, A; Martin, R; Ramaswami, U; Gucsavas-Calikoglu, M; Vijayaraghavan, S; Wendt, S; Puga, AC; Ulbrich, B; Shinawi, M; Cleary, M; Piper, D; Conway, AM; Kimura, A (August 2006). "A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome)". Genetics in Medicine. 8 (8): 465–73. doi:10.1097/01.gim.0000232477.37660.fb. PMID 16912578.
- Guffon, N (May 2009). "Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years". 154 (5). Journal of Pediatrics. pp. 733–737. doi:10.1016/j.jpeds.2008.11.041. PMID 19167723.
- Annibali, R (Oct 2013). "Hunter syndrome (Mucopolysaccharidosis type II), severe phenotype: long term follow-up on patients undergone to hematopoietic stem cell transplantation". 65 (5). Minerva Pediatrica. pp. 487–496. PMID 24056375.
- Marchione, Marilyn (7 February 2019). "Tests suggest scientists achieved 1st 'in body' gene editing". AP News. Retrieved 7 February 2019.
- Staff (2 February 2019). "Ascending Dose Study of Genome Editing by the Zinc Finger Nuclease (ZFN) Therapeutic SB-913 in Subjects With MPS II". ClinicalTrials.gov. U.S. National Library of Medicine. Retrieved 7 February 2019.
- LaTercera.com (in Spanish)
- Young ID, Harper PS (1982). "Incidence of Hunter's syndrome". Hum. Genet. 60 (4): 391–2. doi:10.1007/BF00569230. PMID 6809596.
- Hunter's syndrome (Charles A. Hunter) at Who Named It?
- Hunter, C. A. (1917). "A Rare Disease in Two Brothers". Proceedings of the Royal Society of Medicine. London. 10 (Sect Study Dis Child): 104–116. PMC 2018097. PMID 19979883.
- "A Phase I/II, Randomized, Safety and Ascending Dose Ranging Study of Intrathecal Idursulfase-IT Administered in Conjunction With Intravenous Elaprase in Pediatric Patients With Hunter Syndrome and Cognitive Impairment". Clinicaltrials.gov. U.S. National Institutes of Health. 15 June 2009. Retrieved 22 July 2018.
- "A Safety and Dose Ranging Study of Idursulfase (Intrathecal) Administration Via an Intrathecal Drug Delivery Device in Pediatric Patients With Hunter Syndrome Who Have Central Nervous System Involvement and Are Receiving Treatment With Elaprase® - Results". Clinicaltrials.gov. U.S. National Institutes of Health. 2013-10-31. Retrieved 2014-07-20.
- "Study of Intrathecal Idursulfase-IT Administered in Conjunction With Elaprase® in Pediatric Patients With Hunter Syndrome and Early Cognitive Impairment (AIM-IT)". Clinicaltrials.gov. U.S. National Institutes of Health. July 2014. Retrieved 2014-07-20.
- Marchione, Marilynn (15 November 2017). "US scientists try 1st gene editing in the body". Associated Press. Retrieved 16 November 2017.
- Marchione, Marilynne (2017-11-14). "Scientists Attempt First Gene Editing Inside a Patient". Time. Retrieved 2017-11-15.
- Marchione, Mailynn (2018-09-05). "Early results boost hopes for historic gene editing attempt". AP News. Retrieved 2018-09-06.
- NEWS.BBC.co.uk, "Father cleared of murdering son", BBC News
- Guardian.co.uk, "Former SAS soldier who smothered terminally ill son walks free" The Guardian
- NEWS.BBC.co.uk, "Review 'will clarify murder laws'" BBC News
External links

- GeneReview/NIH/UW entry on Mucopolysaccharidosis Type II
| Classification | |
|---|---|
| External resources |