HFE hereditary haemochromatosis
Hereditary haemochromatosis (or hemochromatosis)[3] is a genetic disorder characterized by excessive intestinal absorption of dietary iron, resulting in a pathological increase in total body iron stores.[4] Humans, like most animals, have no means to excrete excess iron.[5]
| Haemochromatosis type 1 | |
|---|---|
| Other names | HFE hereditary haemochromatosis[1] HFE-related hereditary haemochromatosis[2] |
| Specialty | Endocrinology, hepatology |
Excess iron accumulates in tissues and organs, disrupting their normal function. The most susceptible organs include the liver, adrenal glands, heart, skin, gonads, joints, and the pancreas; patients can present with cirrhosis, polyarthropathy, adrenal insufficiency, heart failure, or diabetes.[6]
The hereditary form of the disease is most common among those of Northern European ancestry, in particular those of Celtic descent.[7] The disease is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations.[8] Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.[8]
Signs and symptoms
Haemochromatosis is protean in its manifestations, i.e., often presenting with signs or symptoms suggestive of other diagnoses that affect specific organ systems. Many of the signs and symptoms below are uncommon, and most patients with the hereditary form of haemochromatosis do not show any overt signs of disease nor do they suffer premature morbidity, if they are diagnosed early, but more often than not, diagnosis occurs in the autopsy [9]
Presently, the classic triad of cirrhosis, bronze skin, and diabetes is less common because of earlier diagnosis.[10]
The more common clinical manifestations include:[6][10][11][12]
- Fatigue
- Malaise
- Joint and bone pain
- Liver cirrhosis (with an increased risk of hepatocellular carcinoma): Liver disease is always preceded by evidence of liver dysfunction, including elevated serum enzymes specific to the liver, clubbing of the fingers, leuconychia, asterixis, hepatomegaly, palmar erythema, and spider naevi. Cirrhosis can also present with jaundice (yellowing of the skin) and ascites.
- Insulin resistance (often, patients have already been diagnosed with diabetes mellitus type 2) due to pancreatic damage from iron deposition.)
- Erectile dysfunction and hypogonadism, resulting in decreased libido
- Congestive heart failure, abnormal heart rhythms, or pericarditis
- Arthritis of the hands (especially the second and third metacarpophalangeal joints), but also the knee and shoulder joints
- Damage to the adrenal gland, leading to adrenal insufficiency
Less common findings including:
- Deafness[13]
- Dyskinesias, including Parkinsonian symptoms[13][14][15]
- Dysfunction of certain endocrine organs:
- Parathyroid gland (leading to hypocalcaemia)
- Pituitary gland
- More commonly, a slate-grey or less commonly darkish colour to the skin (see pigmentation, hence its name "bronze diabetes" when it was first described by Armand Trousseau in 1865)
- An increased susceptibility to certain infectious diseases caused by siderophilic microorganisms:
- Vibrio vulnificus infections from eating seafood or wound infection[16]
- Listeria monocytogenes
- Yersinia enterocolica
- Salmonella enterica (serotype Typhymurium)[17]
- Klebsiella pneumoniae
- Escherichia coli
- Rhizopus arrhizus
- Mucor species
- Aspergillus fumigatus
- Cytomegalovirus
- Hepatitis B virus
- Hepatitis C virus
Males are usually diagnosed after their forties and fifties, and women several decades later, owing to the fact that symptoms mimic those of menopause. Most people display symptoms in their 30s but due to the lack of knowledge surrounding haemochromatosis, they are diagnosed years later. The severity of clinical disease in the hereditary form varies considerably. Some evidence suggests that hereditary haemochromatosis patients affected with other liver ailments such as hepatitis or alcoholic liver disease suffer worse liver disease than those with either condition alone. Also, juvenile forms of hereditary haemochromatosis present in childhood with the same consequences of iron overload.
End-organ damage
Iron is stored in the liver, pancreas, and heart. Long-term effects of haemochromatosis on these organs can be very serious, even fatal when untreated.[18] For example, similar to alcoholism, haemochromatosis can cause cirrhosis of the liver. The liver is a primary storage area for iron and naturally accumulates excess iron. Over time, the liver is likely to be damaged by iron overload. Cirrhosis itself may lead to additional and more serious complications, including bleeding from dilated veins in the esophagus (esophageal varices) and stomach (gastric varices) and severe fluid retention in the abdomen (ascites). Toxins may accumulate in the blood and eventually affect mental functioning. This can lead to confusion or even coma (hepatic encephalopathy).
Cirrhosis and haemochromatosis together can increase the risk of liver cancer. (Nearly one-third of people with haemochromatosis and cirrhosis eventually develop liver cancer.)
The pancreas, which also stores iron, is very important in the body’s mechanisms for sugar metabolism. Diabetes affects the way the body uses blood sugar (glucose). Diabetes is, in turn, the leading cause of new blindness in adults and may be involved in kidney failure and cardiovascular disease.
If excess iron in the heart interferes with its ability to circulate enough blood, a number of problems can occur, such as congestive heart failure and death. The condition may be reversible when haemochromatosis is treated and excess iron stores are reduced.
Arrhythmia or abnormal heart rhythms can cause heart palpitations, chest pain, and light-headedness, and are occasionally life-threatening. This condition can often be reversed with treatment for haemochromatosis.
Bronze or grey coloration of the skin pigmentation is caused primarily by increased melanin deposition, with iron deposition playing a lesser role.[19]
Severity of periodontal disease is associated with high transferrin saturation in haemochromatosis patients.[20][21]
Genetics
The regulation of dietary iron absorption is complex and understanding is incomplete. One of the better-characterized genes responsible for hereditary haemochromatosis is HFE[22] on chromosome 6, which codes for a protein that participates in the regulation of iron absorption. The HFE gene has three common mutations, C282Y, H63D and S65C.[23] The C282Y allele is a transition point mutation from guanine to adenine at nucleotide 845 in HFE, resulting in a missense mutation that replaces the cysteine residue at position 282 with a tyrosine amino acid.[24] Heterozygotes for either allele can manifest clinical iron overload, if they have two of any alleles. This makes them compound heterozygous for haemochromatosis and puts them greatly at risk of storing excess iron in the body.
Mutations of the HFE gene account for 90% of the cases of nontransfusional iron overload. This gene is closely linked to the HLA-A3 locus. Homozygosity for the C282Y mutation is the most common genotype responsible for clinical iron accumulation, though heterozygosity for C282Y/H63D mutations, so-called compound heterozygotes, results in clinically evident iron overload. Considerable debate exists regarding the penetrance—the probability of clinical expression of the trait given the genotype— for clinical disease in homozygotes. Most, if not all, males homozygous for HFE C282Y show manifestations of liver dysfunction such as elevated liver-specific enzymes such as serum gamma glutamyltransferase by late middle age.
Each patient with the susceptible genotype accumulates iron at different rates depending on iron intake, the exact nature of the mutation, and the presence of other insults to the liver, such as alcohol and viral disease. As such, the degree to which the liver and other organs is affected is highly variable and is dependent on these factors and co-morbidities, as well as age at which they are studied for manifestations of disease.[25] Penetrance differs between populations.
Individuals with the relevant mutations may never develop iron overload. Phenotypic expression is present in 70% of C282Y homozygotes with less than 10% going on to experience severe iron overload and organ damage.[26]
Pathophysiology
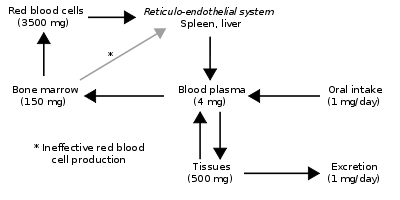
Since the regulation of iron metabolism is still poorly understood, a clear model of how haemochromatosis operates is still not available. A working model describes the defect in the HFE gene, where a mutation puts the intestinal absorption of iron into overdrive. Normally, HFE facilitates the binding of transferrin, which is iron's carrier protein in the blood. Transferrin levels are typically elevated at times of iron depletion (low ferritin stimulates the release of transferrin from the liver). When transferrin is high, HFE works to increase the intestinal release of iron into the blood. When HFE is mutated, the intestines perpetually interpret a strong transferrin signal as if the body were deficient in iron. This leads to maximal iron absorption from ingested foods and iron overload in the tissues. However, HFE is only part of the story, since many patients with mutated HFE do not manifest clinical iron overload, and some patients with iron overload have a normal HFE genotype. A possible explanation is the fact that HFE' normally plays a role in the production of hepcidin in the liver, a function that is impaired in HFE mutations.[27]
People with abnormal iron regulatory genes do not reduce their absorption of iron in response to increased iron levels in the body. Thus, the iron stores of the body increase. As they increase, the iron which is initially stored as ferritin is deposited in organs as haemosiderin and this is toxic to tissue, probably at least partially by inducing oxidative stress.[28] Iron is a pro-oxidant. Thus, haemochromatosis shares common symptomology (e.g., cirrhosis and dyskinetic symptoms) with other "pro-oxidant" diseases such as Wilson's disease, chronic manganese poisoning, and hyperuricaemic syndrome in Dalmatian dogs. The latter also experience "bronzing".
Diagnosis
The diagnosis of haemochromatosis is often made following the incidental finding on routine blood screening of elevated serum liver enzymes or elevation of the transferrin saturation. Arthropathy with stiff joints, diabetes, or fatigue, may be the presenting complaint.[29]
Blood tests
Serum transferrin and transferrin saturation are commonly used as screening for haemochromatosis. Transferrin binds iron and is responsible for iron transport in the blood.[30] Measuring transferrin provides a crude measure of iron stores in the body. Fasting transferrin saturation values in excess of 45% for males or 35% in premenopausal women (i.e. 300 ng/l in males and 200 ng/l in females) are recognized as a threshold for further evaluation of haemochromatosis.[10][31] Transferrin saturation greater than 62% is suggestive of homozygosity for mutations in the HFE gene.[32]
Ferritin, a protein synthesized by the liver, is the primary form of iron storage within cells and tissues. Measuring ferritin provides another crude estimate of whole-body iron stores, though many conditions, particularly inflammation (but also chronic alcohol consumption, nonalcoholic fatty liver disease, and others), can elevate serum ferritin, which can account for up to 90% of cases where elevated levels are observed.[4] Normal values for males are 12–300 ng/ml and for female, 12–150 ng/ml.[29][33] Serum ferritin in excess of 1000 ng/ml of blood is almost always attributable to haemochromatosis.
Other blood tests routinely performed include blood count, renal function, liver enzymes, electrolytes, and glucose (and/or an oral glucose tolerance test).
Liver biopsy

Liver biopsies involve taking a sample of tissue from the liver, using a thin needle. The amount of iron in the sample is then quantified and compared to normal, and evidence of liver damage, especially cirrhosis, is measured microscopically. Formerly, this was the only way to confirm a diagnosis of haemochromatosis, but measures of transferrin and ferritin along with a history are considered adequate in determining the presence of the malady. Risks of biopsy include bruising, bleeding, and infection. Now, when a history and measures of transferrin or ferritin point to haemochromatosis, whether a liver biopsy is still necessary to quantify the amount of accumulated iron is debatable.[29]
MRI
MRI-based testing is a noninvasive and accurate alternative to measure liver iron concentrations.[34]
Other imaging
Clinically, the disease may be silent, but characteristic radiological features may point to the diagnosis. The increased iron stores in the organs involved, especially in the liver and pancreas, result in characteristic findings on unenhanced CT and a decreased signal intensity in MRI scans. Haemochromatosis arthropathy includes degenerative osteoarthritis and chondrocalcinosis. The distribution of the arthropathy is distinctive, but not unique, frequently affecting the second and third metacarpophalangeal joints of the hand. The arthropathy can, therefore, be an early clue as to the diagnosis of haemochromatosis.
Functional testing
Based on the history, the doctor might consider specific tests to monitor organ dysfunction, such as an echocardiogram for heart failure, or blood glucose monitoring for patients with haemochromatosis diabetes.
Stages
The American Association for the Study of Liver Diseases suggests the following three stages for the condition (identified by the European Association for the Study of Liver Diseases):[26]
- Genetic susceptibility but no iron overload. Individuals who have the genetic disorder only.
- Iron overload but no organ or tissue damage.
- Organ or tissue damage as a result of iron deposition.
Individuals at each stage do not necessarily progress on to the next stage, and end stage disease is more common in males.
Differential diagnosis
Other causes of excess iron accumulation exist, which have to be considered before haemochromatosis is diagnosed.
- African iron overload, formerly known as Bantu siderosis, was first observed among people of African descent in Southern Africa. Originally, this was blamed on ungalvanised barrels used to store home-made beer, which led to increased oxidation and increased iron levels in the beer. Further investigation has shown that only some people drinking this sort of beer get an iron overload syndrome, and that a similar syndrome occurred in people of African descent who have had no contact with this kind of beer (e.g., African Americans). This led investigators to the discovery of a gene polymorphism in the gene for ferroportin, which predisposes some people of African descent to iron overload.[35]
- Transfusion haemosiderosis is the accumulation of iron, mainly in the liver, in patients who receive frequent blood transfusions (such as those with thalassaemia).
- Dyserythropoeisis, also known as myelodysplastic syndrome, is a disorder in the production of red blood cells. This leads to increased iron recycling from the bone marrow and accumulation in the liver.
Screening
Standard diagnostic measures for haemochromatosis, transferrin saturation and ferritin tests, are not a part of routine medical testing. Screening for haemochromatosis is recommended if the patient has a parent, child, or sibling with the disease.[36]
Routine screening of the general population for hereditary haemochromatosis is generally not done. Mass genetic screening has been evaluated by the U.S. Preventive Services Task Force, among other groups, which recommended against genetic screening of the general population for hereditary haemochromatosis because the likelihood of discovering an undiagnosed patient with clinically relevant iron overload is less than one in 1,000. Although strong evidence shows that treatment of iron overload can save lives in patients with transfusional iron overload, no clinical study has shown that for asymptomatic carriers of hereditary haemochromatosis treatment with venesection (phlebotomy) provides any clinical benefit.[37][38] Recently, patients are suggested to be screened for iron overload using serum ferritin as a marker. If serum ferritin exceeds 1000 ng/ml, iron overload is very likely the cause.
Treatment
Phlebotomy
Early diagnosis is vital, as the late effects of iron accumulation can be wholly prevented by periodic phlebotomies (by venesection) comparable in volume to blood donations.[39] Initiation of treatment is recommended when ferritin levels reach 500 μg/l.[40]
Phlebotomy (or bloodletting) is usually done at a weekly interval until ferritin levels are less than 50 μg/l. To prevent iron reaccumulation, subsequent phlebotomies are normally carried out about once every three to four months for males, and twice a year for females.[41]
Desferrioxamine mesilate
Where venesection is not possible, long-term administration of desferrioxamine mesylate is useful. Desferrioxamine is an iron-chelating compound, and excretion induced by desferrioxamine is enhanced by administration of vitamin C. It cannot be used during pregnancy or breast-feeding due to risk of defects in the child.
Organ damage
- Treatment of organ damage (heart failure with diuretics and ACE inhibitor therapy)
Diet
- Limiting intake of alcoholic beverages, vitamin C (increases iron absorption in the gut), red meat (high in iron), and potential causes of food poisoning (shellfish, seafood)[42]
- Increasing intake of substances that inhibit iron absorption, such as high-tannin tea, calcium, and foods containing oxalic and phytic acids (such as collard greens, which must be consumed at the same time as the iron-containing foods to be effective)[43]
Prognosis
Persons with symptomatic haemochromatosis have somewhat reduced life expectancy compared to the general population, mainly due to excess mortality from cirrhosis and liver cancer. Patients who were treated with phlebotomy lived longer than those who were not.[44][45] Patients without liver disease or diabetes had similar survival rate to the general population.
Epidemiology
Haemochromatosis is one of the most common heritable genetic conditions in people of Northern European extraction, with a prevalence of one in 200. The disease has a variable penetration, and about one in 10 people of this demographic carry a mutation in one of the genes regulating iron metabolism, the most common allele being the C282Y allele in the HFE gene.[46] The prevalence of mutations in iron-metabolism genes varies in different populations. A study of 3,011 unrelated white Australians found that 14% were heterozygous carriers of an HFE mutation, 0.5% were homozygous for an HFE mutation, and only 0.25% of the study population had clinically relevant iron overload. Most patients who are homozygous for HFE mutations do not manifest clinically relevant haemochromatosis (see Genetics above).[25] Other populations have a lower prevalence of both the genetic mutation and the clinical disease.
Genetics studies suggest the original haemochromatosis mutation arose in a single person, possibly of Celtic ethnicity, who lived 60–70 generations ago.[47] At that time, when dietary iron may have been scarcer than today, the presence of the mutant allele may have provided an evolutionary or natural selection reproductive advantage by maintaining higher iron levels in the blood.
Terminology
The term "haemochromatosis" is used by different sources in many different ways.
It is often used to imply an association with the HFE gene. For many years, HFE was the only known gene associated with haemochromatosis, and the term "hereditary haemochromatosis" was used to describe haemochromatosis type 1. However, many different genetic associations with this condition are now known. The older the text, or the more general the audience, the more likely that HFE is implied.
"Haemochromatosis" has also been used in contexts where a genetic cause for iron accumulation had not been known. In some cases, however, a condition that was thought to be due to diet or environment was later linked to a genetic polymorphism, as in African iron overload.
History
The disease was first described in 1865 by Armand Trousseau in a report on diabetes in patients presenting with a bronze pigmentation of their skin.[48] Trousseau did not associate diabetes with iron accumulation; the recognition that infiltration of the pancreas with iron might disrupt endocrine function resulting in diabetes was made by Friedrich Daniel von Recklinghausen in 1890.[49][50]
References
- Allen KJ, Gurrin LC, Constantine CC, et al. (January 2008). "Iron-overload-related disease in HFE hereditary hemochromatosis" (PDF). N. Engl. J. Med. 358 (3): 221–30. doi:10.1056/NEJMoa073286. PMID 18199861.
- Jacobs EM, Verbeek AL, Kreeftenberg HG, et al. (December 2007). "Changing aspects of HFE-related hereditary haemochromatosis and endeavours to early diagnosis". Neth J Med. 65 (11): 419–24. PMID 18079564.
- Franchini M (March 2006). "Hereditary iron overload: update on pathophysiology, diagnosis, and treatment". Am. J. Hematol. 81 (3): 202–9. doi:10.1002/ajh.20493. PMID 16493621.
- St John, Andrew. "Testing for HFE-related haemochromatosis". Australian Prescriber (34): 73–6. Archived from the original on 2011-07-27. Retrieved 2011-06-03.
- "The interaction of iron and erythropoietin".
- Iron Overload and Hemochromatosis Centers for Disease Control and Prevention
- "News | Penn State University". news.psu.edu.
- Reference, Genetics Home. "Hereditary hemochromatosis". Genetics Home Reference.
- Hemochromatosis-Diagnosis Archived 2007-03-18 at the Wayback Machine National Digestive Diseases Information Clearinghouse, National Institutes of Health, U.S. Department of Health and Human Services
- Pietrangelo A (June 2004). "Hereditary hemochromatosis—a new look at an old disease". N. Engl. J. Med. 350 (23): 2383–97. doi:10.1056/NEJMra031573. PMID 15175440.
- Hemochromatosis Archived 2007-03-18 at the Wayback Machine National Digestive Diseases Information Clearinghouse, National Institutes of Health, U.S. Department of Health and Human Services
- "Hemochromatosis: Symptoms". Mayo Foundation for Medical Education and Research (MFMER).
- Jones H, Hedley-Whyte E (1983). "Idiopathic hemochromatosis (IHC): dementia and ataxia as presenting signs". Neurology. 33 (11): 1479–83. doi:10.1212/WNL.33.11.1479. PMID 6685241.
- Costello D, Walsh S, Harrington H, Walsh C (2004). "Concurrent hereditary haemochromatosis and idiopathic Parkinson's disease: a case report series". J Neurol Neurosurg Psychiatry. 75 (4): 631–3. doi:10.1136/jnnp.2003.027441. PMC 1739011. PMID 15026513.
- Nielsen J, Jensen L, Krabbe K (1995). "Hereditary haemochromatosis: a case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome". J Neurol Neurosurg Psychiatry. 59 (3): 318–21. doi:10.1136/jnnp.59.3.318. PMC 486041. PMID 7673967.
- Barton JC, Acton RT (April 2009). "Hemochromatosis and Vibrio vulnificus Wound Infections". J. Clin. Gastroenterol. 43 (9): 890–893. doi:10.1097/MCG.0b013e31819069c1. PMID 19349902.
- Jolivet-Gougeon A, Loréal O, Ingels A, et al. (October 2008). "Serum transferrin saturation increase is associated with decrease of antibacterial activity of serum in patients with HFE-related genetic hemochromatosis". Am. J. Gastroenterol. 103 (10): 2502–8. doi:10.1111/j.1572-0241.2008.02036.x. PMID 18684194.
- "Hemochromatosis: Complications". Mayo Foundation for Medical Education and Research (MFMER).
- Pedrup A, Poulsen H (1964). "Hemochromatosis and Vitiligo". Archives of Dermatology. 90 (1): 34–37. doi:10.1001/archderm.1964.01600010040010. PMID 14149720.
- Meuric, Vincent; Lainé, Fabrice; Boyer, Emile; Le Gall-David, Sandrine; Oger, Emmanuel; Bourgeois, Denis; Bouchard, Philippe; Bardou-Jacquet, Edouard; Turmel, Valérie (September 2017). "Periodontal status and serum biomarker levels in HFE haemochromatosis patients. A case-series study". Journal of Clinical Periodontology. 44 (9): 892–897. doi:10.1111/jcpe.12760. ISSN 1600-051X. PMID 28586532.
- Boyer, Emile; Le Gall-David, Sandrine; Martin, Bénédicte; Fong, Shao Bing; Loréal, Olivier; Deugnier, Yves; Bonnaure-Mallet, Martine; Meuric, Vincent (2018-10-19). "Increased transferrin saturation is associated with subgingival microbiota dysbiosis and severe periodontitis in genetic haemochromatosis". Scientific Reports. 8 (1): 15532. Bibcode:2018NatSR...815532B. doi:10.1038/s41598-018-33813-0. ISSN 2045-2322. PMC 6195524. PMID 30341355.
- Olynyk JK, Trinder D, Ramm GA, Britton RS, Bacon BR (September 2008). "Hereditary hemochromatosis in the post-HFE era". Hepatology. 48 (3): 991–1001. doi:10.1002/hep.22507. PMC 2548289. PMID 18752323.
- "Hemochromatosis: Causes". Mayo Foundation for Medical Education and Research (MFMER).
- Feder JN, Gnirke A, Thomas W, et al. (1996). "A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis". Nature Genetics. 13 (4): 399–408. doi:10.1038/ng0896-399. PMID 8696333.
- Olynyk J, Cullen D, Aquilia S, Rossi E, Summerville L, Powell L (1999). "A population-based study of the clinical expression of the hemochromatosis gene" (PDF). N Engl J Med. 341 (10): 718–24. doi:10.1056/NEJM199909023411002. PMID 10471457.
- Bacon, Bruce R.; Adams, Paul C.; Kowdley, Kris V.; Powell, Lawrie W.; Tavill, Anthony S. (July 2011). "Diagnosis and management of hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases". Hepatology. 54 (1): 328–343. doi:10.1002/hep.24330. PMC 3149125. PMID 21452290.
- Vujić Spasić M, Kiss J, Herrmann T, et al. (2008). "Hfe acts in hepatocytes to prevent hemochromatosis". Cell Metab. 7 (2): 173–8. doi:10.1016/j.cmet.2007.11.014. PMID 18249176.
- Shizukuda Y, Bolan C, Nguyen T, Botello G, Tripodi D, Yau Y, Waclawiw M, Leitman S, Rosing D (2007). "Oxidative stress in asymptomatic subjects with hereditary hemochromatosis". Am J Hematol. 82 (3): 249–50. doi:10.1002/ajh.20743. PMID 16955456.
- "Hemochromatosis: Tests and diagnosis". Mayo Foundation for Medical Education and Research (MFMER). Retrieved 2009-04-20.
- "Transerrin and Iron Transport Physiology". sickle.bwh.harvard.edu. Archived from the original on 2007-03-07. Retrieved 2007-03-17.
- Adams, PC; Reboussin, DM; Barton, JC; McLaren, CE; Eckfeldt, JH; McLaren, GD; Dawkins, FW; Acton, RT; Harris, EL; Gordeuk, VR; Leiendecker-Foster, C; Speechley, M; Snively, BM; Holup, JL; Thomson, E; Sholinsky, P; Hemochromatosis and Iron Overload Screening (HEIRS) Study Research, Investigators (28 April 2005). "Hemochromatosis and iron-overload screening in a racially diverse population". The New England Journal of Medicine. 352 (17): 1769–78. doi:10.1056/nejmoa041534. PMID 15858186.
- Dadone MM, Kushner JP, Edwards CQ, Bishop DT, Skolnick MH (August 1982). "Hereditary hemochromatosis. Analysis of laboratory expression of the disease by genotype in 18 pedigrees". American Journal of Clinical Pathology. 78 (2): 196–207. doi:10.1093/ajcp/78.2.196. PMID 7102818.
- MedlinePlus Encyclopedia Ferritin Test Measuring iron in the body
- St Pierre; Clark, PR; Chua-Anusorn, W; Fleming, AJ; Jeffrey, GP; Olynyk, JK; Pootrakul, P; Robins, E; Lindeman, R (2005). "Non-invasive measurement and imaging of liver iron concentrations using proton magnetic resonance". Blood. 105 (2): 855–61. doi:10.1182/blood-2004-01-0177. PMID 15256427.
- Gordeuk V, Caleffi A, Corradini E, Ferrara F, Jones R, Castro O, Onyekwere O, Kittles R, Pignatti E, Montosi G, Garuti C, Gangaidzo I, Gomo Z, Moyo V, Rouault T, MacPhail P, Pietrangelo A (2003). "Iron overload in Africans and African-Americans and a common mutation in the SCL40A1 (ferroportin 1) gene". Blood Cells Mol Dis. 31 (3): 299–304. doi:10.1016/S1079-9796(03)00164-5. PMID 14636642.
- "Summaries for patients. Screening for hereditary hemochromatosis: recommendations from the American College of Physicians". Ann. Intern. Med. 143 (7): I-46. 2005. doi:10.7326/0003-4819-143-7-200510040-00004. PMID 16204158.
- U.S. Preventive Services Task Force (2006). "Screening for haemochromatosis: recommendation statement". Ann. Intern. Med. 145 (3): 204–8. doi:10.7326/0003-4819-145-3-200608010-00008. PMID 16880462.
- Screening for Hemochromatosis U.S. Preventive Services Task Force (2006). Summary of Screening Recommendations and Supporting Documents. Retrieved 18 March 2007
- "Hemochromatosis: Treatments and drugs". Mayo Foundation for Medical Education and Research (MFMER).
- European Association For The Study Of The Liver. (2010). "EASL clinical practice guidelines for HFE hemochromatosis". Journal of Hepatology. 53 (1): 3–22. doi:10.1016/j.jhep.2010.03.001. PMID 20471131.
- Kowdley, KV; Bennett, RL; Motulsky, AG; Pagon, RA; Adam, MP; Ardinger, HH; Wallace, SE; Amemiya, A; Bean, LJH; Bird, TD; Dolan, CR; Fong, CT; Smith, RJH; Stephens, K (1993). "HFE-Associated Hereditary Hemochromatosis". PMID 20301613. Retrieved 25 May 2015. Cite journal requires
|journal=(help) - Plaut, David; McLellan, William (2009). "Hereditary hemochromatosis". Journal of Continuing Education Topics & Issues. 11 (1). Archived from the original on 2016-10-11. Retrieved 11 October 2016.
- http://dynaweb.ebscohost.com/Detail.aspx?id=116469&sid=14aa79e5-a881-407c-94e7-339b81c4cd18@sessionmgr3%5B%5D accessed October 15, 2008.
- Niederau, Claus; Fischer, Rudolf; Sonnenberg, Amnon; Stremmel, Wolfgang; Trampisch, Hans J.; Strohmeyer, Georg (1985-11-14). "Survival and Causes of Death in Cirrhotic and in Noncirrhotic Patients with Primary Hemochromatosis". New England Journal of Medicine. 313 (20): 1256–1262. doi:10.1056/NEJM198511143132004. ISSN 0028-4793. PMID 4058506.
- Bokhoven, M. A. van; Deursen, C. Th B. M. van; Swinkels, D. W. (2011-01-19). "Diagnosis and management of hereditary haemochromatosis". BMJ. 342: c7251. doi:10.1136/bmj.c7251. hdl:2066/95805. ISSN 0959-8138. PMID 21248018.
- Mendes AI, Ferro A, Martins R, et al. (March 2009). "Non-classical hereditary hemochromatosis in Portugal: novel mutations identified in iron metabolism-related genes" (PDF). Ann. Hematol. 88 (3): 229–34. doi:10.1007/s00277-008-0572-y. PMID 18762941.
- "Archived copy" (PDF). Archived from the original (PDF) on 2008-12-02. Retrieved 2014-01-07.CS1 maint: archived copy as title (link)
- Trousseau A (1865). "Glycosurie, diabète sucré". Clinique Médicale de l'Hôtel-Dieu de Paris. 2: 663–98.
- von Recklinghausen FD (1890). "Hämochromatose". Tageblatt der Naturforschenden Versammlung 1889: 324.
- "Friedrich Daniel von Recklinghausen". www.whonamedit.com.
External links
| Classification | |
|---|---|
| External resources |