Early-onset Alzheimer's disease
Early-onset Alzheimer's disease, also called early-onset Alzheimer's, or early-onset AD, is Alzheimer's disease diagnosed before the age of 65. It is an uncommon form of Alzheimer's, accounting for only 5-10% of all Alzheimer's cases. About 13% of the cases of early-onset Alzheimer's are familial,[1] where a genetic predisposition leads to the disease. The other incidences of early-onset Alzheimer's, however, share the same traits as the "late-onset" form of Alzheimer's disease, and little is understood about how it starts.
| Early-onset Alzheimer's disease | |
|---|---|
| Specialty | Neurology |
Nonfamilial early-onset AD can develop in people who are in their 30s or 40s, but this is extremely rare.[2] The majority of people with early-onset Alzheimer's are in their 50s or early 60s.
History of Alzheimer's disease
The symptoms of the disease as a distinct nosologic entity were first identified by Emil Kraepelin, and the characteristic neuropathology was first observed by Alois Alzheimer in 1906. In this sense, the disease was co-discovered by Kraepelin and Alzheimer, who worked in Kraepelin's laboratory. Because of the overwhelming importance Kraepelin attached to finding the neuropathological basis of psychiatric disorders, Kraepelin made the decision that the disease would bear Alzheimer's name.[3]
Familial Alzheimer's disease
Familial Alzheimer's disease (FAD) or early-onset familial Alzheimer's disease (EOFAD) is an uncommon form of Alzheimer's disease that usually strikes earlier in life, defined as before the age of 65 (usually between 50 and 65 years of age) and is inherited in an autosomal dominant fashion, identified by genetics and other characteristics such as the age of onset. Familial AD requires the patient to have at least one first-degree relative with a history of AD. Nonfamilial cases of AD are referred to as "sporadic" AD, where genetic risk factors are minor or unclear.
While early-onset familial AD is estimated to account for only 3.5% of total Alzheimer's disease,[2] it has presented a useful model in studying various aspects of the disorder. Currently, the early-onset familial AD gene mutations guide the vast majority of animal model-based therapeutic discovery and development for AD.
Clinical features
Alzheimer's disease (AD) is the most common cause of dementia and usually occurs in old age. It is invariably fatal, generally within 10 years of the first signs. Early signs of AD include unusual memory loss, particularly in remembering recent events and the names of people and things, logopenic primary progressive aphasia. As the disease progresses, the patient exhibits more serious problems, becoming subject to mood swings and unable to perform complex activities such as driving. In the latter stages, they forget how to do simple things such as brushing their hair and then require full-time care.
Histologically, familial AD is practically indistinguishable from other forms of the disease. Deposits of amyloid can be seen in sections of brain tissue. This amyloid protein forms plaques and neurofibrillary tangles that progress through the brain. Very rarely, the plaque may be unique, or uncharacteristic of AD; this can happen when a mutation occurs in one of the genes that creates a functional, but malformed, protein instead of the ineffective gene products that usually result from mutations.
The underlying neurobiology of this disease is just recently starting to be understood. Researchers have been working on mapping the inflammation pathways associated with the development, progression, and degenerative properties of AD. The major molecules involved in these pathways include glial cells (specifically astrocytes and microglia), beta-amyloid, and proinflammatory compounds.
Beta-amyloid is a small piece of a larger protein called the amyloid precursor protein (APP). Once APP is activated, it is cut into smaller sections of other proteins. One of the fragments produced in this cutting process is β-amyloid. β-amyloid is “stickier” than any other fragment produced from cut-up APP, so it starts an accumulation process in the brain, which is due to various genetic and biochemical abnormalities. Eventually, the fragments form oligomers, then fibrils, beta-sheets, and finally plaques. The presence of β-amyloid plaques in the brain causes the body to recruit and activate microglial cells and astrocytes.
Genetics
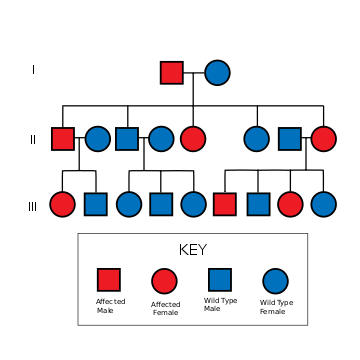
Familial Alzheimer disease is caused by a mutation in one of at least three genes, which code for presenilin 1, presenilin 2, and amyloid precursor protein (APP).[4][5][6] Other gene mutations are in study.
PSEN1 – Presenilin 1
The presenilin 1 gene (PSEN1 located on chromosome 14) was identified by Sherrington (1995)[7] and multiple mutations have been identified. Mutations in this gene cause familial Alzheimer's type 3 with certainty and usually under 50 years old. This protein has been identified as part of the enzymatic complex that cleaves amyloid beta peptide from APP (see below).
The gene contains 14 exons, and the coding portion is estimated at 60 kb, as reported by Rogaev (1997)[8] and Del-Favero (1999).[9] The protein the gene codes for (PS1) is an integral membrane protein. As stated by Ikeuchi (2002)[10] it cleaves the protein Notch1 so is thought by Koizumi (2001)[11] to have a role in somitogenesis in the embryo. It also has an action on an amyloid precursor protein, which gives its probable role in the pathogenesis of FAD. Homologs of PS1 have been found in plants, invertebrates and other vertebrates.
Some of the mutations in the gene, of which over 90 are known, include: His163Arg, Ala246Glu, Leu286Val and Cys410Tyr. Most display complete penetrance, but a common mutation is Glu318Gly and this predisposes individuals to familial AD, with a study by Taddei (2002)[12] finding an incidence of 8.7% in patients with familial AD.
PSEN2 – Presenilin 2
The presenilin 2 gene (PSEN2) is very similar in structure and function to PSEN1. It is located on chromosome 1 (1q31-q42), and mutations in this gene cause type 4 FAD. The gene was identified by Rudolph Tanzi and Jerry Schellenberg in 1995.[13] A subsequent study by Kovacs (1996)[14] showed that PS1 and PS2 proteins are expressed in similar amounts, and in the same organelles as each other, in mammalian neuronal cells. Levy-Lahad (1996)[15] determined that PSEN2 contained 12 exons, 10 of which were coding exons, and that the primary transcript encodes a 448-amino-acid polypeptide with 67% homology to PS1. This protein has been identified as part of the enzymatic complex that cleaves amyloid beta peptide from APP (see below).
The mutations have not been studied as much as PSEN1, but distinct allelic variants have been identified. These include Asn141Ile, which was identified first by Rudolph Tanzi and Jerry Schellenberg in Volga German families with familial Alzheimer disease (Levy-Lahad et al. Nature, 1995). One of these studies by Nochlin (1998) found severe amyloid angiopathy in the affected individuals in a family. This phenotype may be explained by a study by Tomita (1997)[16] suggesting that the Asn141Ile mutation alters amyloid precursor protein (APP) metabolism causing an increased rate of protein deposition into plaques.
Other allelic variants are Met239Val which was identified in an Italian pedigree by Rogaev (1995)[17] who also suggested early on that the gene may be similar to PSEN1, and an Asp439Ala mutation in exon 12 of the gene which is suggest by Lleo (2001)[18] to change the endoproteolytic processing of the PS2.
APP – amyloid beta (A4) precursor protein
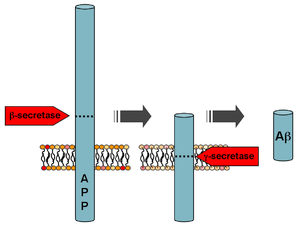
Mutations to the amyloid beta A4 precursor protein (APP) located on the long arm of chromosome 21 (21q21.3) cause familial Alzheimer disease.[6]
Two of the different APP mutations identified and characterized are the Swedish mutation[20] and the Arctic mutation[21]. Functional analyses of these mutations have significantly increased the understanding of the disease pathogenesis. Whereas the Swedish mutation located at the cleavage site for β-secretase, results in an overall higher production of Aβ peptides by increasing the β-secretory cleavage [22], the Arctic mutation leads to a conformation change of the Aβ peptide and increased formation of toxic Aβ protofibrils[23].
Pathophysiology
Following cleavage by β-secretase, APP is cleaved by a membrane-bound protein complex called γ-secretase to generate Aβ. Presenilins 1 and 2 are the enzymatic centers of this complex along with nicastrin, Aph1, and PEN-2. Alpha-secretase cleavage of APP, which precludes the production of Aβ, is the most common processing event for APP. 21 allelic mutations have been discovered in the APP gene. These guarantee onset of early-onset familial Alzheimer disease and all occur in the region of the APP gene that encodes the Aβ domain.
Genetic testing
Genetic testing is available for symptomatic individuals and asymptomatic relatives.[5]
Impact of early-onset Alzheimer's
The atypical lifecourse timing of early-onset Alzheimer's means that it presents distinctive impacts upon experience. For example, the disease can have devastating effects on the careers, caretakers and family members of patients.[24][25]
Those who are working lose their ability to perform their jobs competently, and are forced into early retirement. When this can be predicted, employees must discuss their future with their employers and the loss of skills they expect to face.[26] Those who are forced to retire early may not have access to the full range of benefits available to those who retire at the minimum age set by the government.[26] With some jobs, a mistake may have devastating consequences on a large number of people, and cases have been reported in which a person with early-onset Alzheimer's who is unaware of their condition has caused distress.[27]
Younger people with Alzheimer's may also lose their ability to take care of their own needs, such as money management.[28]
It has also been highlighted, however, that conceptualizations of Alzheimer's and ageing should resist the notion that there are two distinct conditions.[29] A binary model, which focuses in particular on the needs of younger people, could lead to the challenges experienced by older people being understated.[30]
See also
- Still Alice (novel) and the movie Still Alice, whose main protagonist has EOAD
- Spirit Unforgettable, a documentary film about the farewell tour of musician John Mann and his band Spirit of the West following his diagnosis with early-onset Alzheimer's
References
- Campion, Dominique; Dumanchin, Cécile; Hannequin, Didier; Dubois, Bruno; Belliard, Serge; Puel, Michèle; Thomas-Anterion, Catherine; Michon, Agnès; Martin, Cosette; Charbonnier, Françoise; Raux, Grégory; Camuzat, Agnès; Penet, Christiane; Mesnage, Valérie; Martinez, Maria; Clerget-Darpoux, Françoise; Brice, Alexis; Frebourg, Thierry (1999). "Early-Onset Autosomal Dominant Alzheimer Disease: Prevalence, Genetic Heterogeneity, and Mutation Spectrum". The American Journal of Human Genetics. 65 (3): 664–70. doi:10.1086/302553. PMC 1377972. PMID 10441572.
- Harvey, R J (2003). "The prevalence and causes of dementia in people under the age of 65 years". Journal of Neurology, Neurosurgery & Psychiatry. 74 (9): 1206–9. doi:10.1136/jnnp.74.9.1206. PMC 1738690. PMID 12933919.
- Weber, Matthias M. (1997). "Aloys Alzheimer, a coworker of Emil Kraepelin". Journal of Psychiatric Research. 31 (6): 635–43. doi:10.1016/S0022-3956(97)00035-6. PMID 9447568.
- Bertram, Lars; Tanzi, Rudolph E. (2008). "Thirty years of Alzheimer's disease genetics: The implications of systematic meta-analyses". Nature Reviews Neuroscience. 9 (10): 768–78. doi:10.1038/nrn2494. PMID 18802446.
- Williamson, Jennifer; Goldman, Jill; Marder, Karen S. (2009). "Genetic Aspects of Alzheimer Disease". The Neurologist. 15 (2): 80–6. doi:10.1097/NRL.0b013e318187e76b. PMC 3052768. PMID 19276785.
- Ertekin-Taner, Nilüfer (2007). "Genetics of Alzheimer's Disease: A Centennial Review". Neurologic Clinics. 25 (3): 611–67, v. doi:10.1016/j.ncl.2007.03.009. PMC 2735049. PMID 17659183.
- Sherrington, R.; Rogaev, E. I.; Liang, Y.; Rogaeva, E. A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; Tsuda, T.; Mar, L.; Foncin, J.-F.; Bruni, A. C.; Montesi, M. P.; Sorbi, S.; Rainero, I.; Pinessi, L.; Nee, L.; Chumakov, I.; Pollen, D.; Brookes, A.; Sanseau, P.; Polinsky, R. J.; Wasco, W.; Da Silva, H. A. R.; Haines, J. L.; Pericak-Vance, M. A.; Tanzi, R. E.; et al. (1995). "Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease". Nature. 375 (6534): 754–60. Bibcode:1995Natur.375..754S. doi:10.1038/375754a0. PMID 7596406.
- Rogaev, E.I.; Sherrington, R.; Wu, C.; Levesque, G.; Liang, Y.; Rogaeva, E.A.; Ikeda, M.; Holman, K.; Lin, C.; Lukiw, W.J.; De Jong, P.J.; Fraser, P.E.; Rommens, J.M.; St George-Hyslop, P. (1997). "Analysis of the 5′ Sequence, Genomic Structure, and Alternative Splicing of thepresenilin-1Gene (PSEN1) Associated with Early Onset Alzheimer Disease". Genomics. 40 (3): 415–24. doi:10.1006/geno.1996.4523. PMID 9073509.
- Del-Favero, Jurgen; Goossens, Dirk; Van Den Bossche, Dirk; Van Broeckhoven, Christine (1999). "YAC fragmentation with repetitive and single-copy sequences: Detailed physical mapping of the presenilin 1 gene on chromosome 14". Gene. 229 (1–2): 193–201. doi:10.1016/S0378-1119(99)00023-2. PMID 10095119.
- Ikeuchi, Takeshi; Sisodia, Sangram S. (2002). "Cell-Free Generation of the Notch1 Intracellular Domain (NICD) and APP-CTFγ". NeuroMolecular Medicine. 1 (1): 43–54. doi:10.1385/NMM:1:1:43. PMID 12025815.
- Koizumi, K; Nakajima, M; Yuasa, S; Saga, Y; Sakai, T; Kuriyama, T; Shirasawa, T; Koseki, H (2001). "The role of presenilin 1 during somite segmentation". Development. 128 (8): 1391–402. PMID 11262239.
- Taddei, K; Fisher, C; Laws, S M; Martins, G; Paton, A; Clarnette, R M; Chung, C; Brooks, W S; Hallmayer, J; Miklossy, J; Relkin, N; St George-Hyslop, P H; Gandy, S E; Martins, R N (2002). "Association between presenilin-1 Glu318Gly mutation and familial Alzheimer's disease in the Australian population". Molecular Psychiatry. 7 (7): 776–81. doi:10.1038/sj.mp.4001072. PMID 12192622.
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.; Oshima, J.; Pettingell, W.; Yu, C.; Jondro, P.; Schmidt, S.; Wang, K.; Al., e. (1995). "Candidate gene for the chromosome 1 familial Alzheimer's disease locus". Science. 269 (5226): 973–7. Bibcode:1995Sci...269..973L. doi:10.1126/science.7638622. PMID 7638622.
- Kovacs, Dora M.; Fausett, Hillary J.; Page, Keith J.; Kim, Tae-Wan; Moir, Robert D.; Merriam, David E.; Hollister, Richard D.; Hallmark, Olivia G.; Mancini, Ronald; Felsenstein, Kevin M.; Hyman, Bradley T.; Tanzi, Rudolph E.; Wasco, Wilma (1996). "Alzheimer–associated presenilins 1 and 2 : Neuronal expression in brain and localization to intracellular membranes in mammalian cells". Nature Medicine. 2 (2): 224–9. doi:10.1038/nm0296-224. PMID 8574969.
- Levy-Lahad, Ephrat; Poorkaj, Parvoneh; Wang, Kai; Fu, Ying Hui; Oshima, Junko; Mulligan, John; Schellenberg, Gerard D. (1996). "Genomic Structure and Expression of STM2, the Chromosome 1 Familial Alzheimer Disease Gene". Genomics. 34 (2): 198–204. doi:10.1006/geno.1996.0266. PMID 8661049.
- Tomita, T.; Maruyama, K.; Saido, T. C.; Kume, H.; Shinozaki, K.; Tokuhiro, S.; Capell, A.; Walter, J.; Grunberg, J.; Haass, C.; Iwatsubo, T.; Obata, K. (1997). "The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein ending at the 42nd (or 43rd) residue". Proceedings of the National Academy of Sciences. 94 (5): 2025–30. Bibcode:1997PNAS...94.2025T. doi:10.1073/pnas.94.5.2025. JSTOR 41579. PMC 20036. PMID 9050898.
- Rogaev, E. I.; Sherrington, R.; Rogaeva, E. A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; Mar, L.; Sorbi, S.; Nacmias, B.; Piacentini, S.; Amaducci, L.; Chumakov, I.; Cohen, D.; Lannfelt, L.; Fraser, P. E.; Rommens, J. M.; George-Hyslop, P. H. St (1995). "Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene". Nature. 376 (6543): 775–8. Bibcode:1995Natur.376..775R. doi:10.1038/376775a0. PMID 7651536.
- Lleo, A.; Blesa, R.; Gendre, J.; Castellvi, M.; Pastor, P.; Queralt, R.; Oliva, R. (2001). "A novel presenilin 2 gene mutation (D439A) in a patient with early-onset Alzheimer's disease". Neurology. 57 (10): 1926–8. doi:10.1212/WNL.57.10.1926. PMID 11723295.
- Malenka, Eric J. Nestler, Steven E. Hyman, Robert C. (2009). Molecular neuropharmacology : a foundation for clinical neuroscience (2nd ed.). New York: McGraw-Hill Medical. ISBN 9780071481274.
- Mullan, Mike; Crawford, Fiona; Axelman, Karin; Houlden, Henry; Lilius, Lena; Winblad, Bengt; Lannfelt, Lars (August 1992). "A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N–terminus of β–amyloid". Nature Genetics. 1 (5): 345–347. doi:10.1038/ng0892-345. PMID 1302033.
- Nilsberth, Camilla; Westlind-Danielsson, Anita; Eckman, Christopher B.; Condron, Margaret M.; Axelman, Karin; Forsell, Charlotte; Stenh, Charlotte; Luthman, Johan; Teplow, David B. (2001). "The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation" (PDF). Nature Neuroscience. 4 (9): 887–893. doi:10.1038/nn0901-887. PMID 11528419.
- Johnston, Janet A.; Cowburn, Richard F.; Norgren, Svante; Wiehager, Birgitta; Venizelos, Nikolaos; Winblad, Bengt; Vigo-Pelfrey, Carmen; Schenk, Dale; Lannfelt, Lars (1994-11-14). "Increased β-amyloid release and levels of amyloid precursor protein (APP) in fibroblast cell lines from family members with the Swedish Alzheimer's disease APP670/671 mutation". FEBS Letters. 354 (3): 274–278. doi:10.1016/0014-5793(94)01137-0. ISSN 1873-3468. PMID 7957938.
- Johansson, Ann-Sofi; Berglind-Dehlin, Fredrik; Karlsson, Göran; Edwards, Katarina; Gellerfors, Pär; Lannfelt, Lars (2006-06-01). "Physiochemical characterization of the Alzheimer's disease-related peptides Aβ1–42Arctic and Aβ1–42wt". FEBS Journal. 273 (12): 2618–2630. doi:10.1111/j.1742-4658.2006.05263.x. ISSN 1742-4658. PMID 16817891.
- Mayo Clinic staff, Early-onset Alzheimer's: When symptoms begin before 65, Mayo Clinic
- Mary Brophy Marcus, Family shares journey after early Alzheimer's diagnosis, USA Today (September 2, 2008).
- Living With Early-Onset Alzheimer's Disease Archived 2007-10-19 at the Wayback Machine, Cleveland Clinic Health System
- Early Onset Alzheimer's On The Rise, CBS News (March 8, 2008).
- Kathleen Fackelmann, Who thinks of Alzheimer's in someone so young?, USA Today (June 11, 2007).
- Rahman, S. (2016). Young Onset Dementia: a label too far?, Dementia Society, July 27, 2016
- Tolhurst, E. (2016). The Burgeoning Interest in Young Onset Dementia: Redressing the balance or reinforcing ageism?, The International Journal of Ageing and Later Life, 10(2): 9-29.
External links
| Classification |
|---|
- Early-Onset Familial Alzheimer Disease - by Thomas D Bird, MD at GeneRevies (NIH.gov)