Eteplirsen
Eteplirsen (brand name Exondys 51) is a medication to treat, but not cure, some types of Duchenne muscular dystrophy (DMD), caused by a specific mutation. Eteplirsen only targets specific mutations and can be used to treat about 14% of DMD cases.[1][2]
 | |
| Clinical data | |
|---|---|
| Trade names | Exondys 51 |
| Other names | AVI-4658 |
| Routes of administration | Intravenous infusion |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
IUPAC name
| |
| CAS Number | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
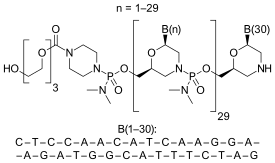
| Formula | C364H569N177O122P30 |
| Molar mass | 10305.886 g·mol−1 |
SMILES
| |
InChI
| |
Eteplirsen was designed and developed by Sarepta Therapeutics. After a controversial debate surrounding the efficacy of the drug, during which two FDA review panel members resigned in protest, eteplirsen received accelerated approval from the US Food and Drug administration in late 2016.[3][4] A year's worth of treatment is priced at $300,000.[5][6]
Adverse effects
The following adverse events were observed in at least 10% of people who received eteplirsen in trials: vomiting, contusion, excoriation, arthralgia, rash, catheter site pain, and upper respiratory tract infection.[7]
Mechanism of action
Duchenne muscular dystrophy is caused when a mutation in the DMD gene changes the DMD RNA so that it no longer codes for functional dystrophin protein, usually due to a mutation that alters the reading frame of the RNA downstream of the mutation. If an exon with an appropriate number of bases lies near the mutation, by removing that exon the downstream reading frame can be corrected and production of partially functional dystrophin can be restored. This is the general strategy used for designing exon-skipping oligos for DMD; as there are 79 exons in the longest splice form of the dystrophin transcript, many different oligos are needed to address the range of mutations present in the population of people with DMD.
Eteplirsen is a morpholino antisense oligomer which triggers excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[8] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (although modified by having an internal deletion consisting of both the patient's original defect, as well as the therapeutically skipped exon) dystrophin.[9] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Exon skipping is induced by eteplirsen, a charge-neutral, phosphorodiamidate morpholino oligomer (PMO) that selectively binds to exon 51 of dystrophin pre-mRNA, restoring the phase of the reading frame and enabling production of functional, but truncated, dystrophin.[10] The uncharged nature of the PMO helps make it resistant to biological degradation.[11] This truncated dystrophin protein produced by eteplirsen causes a less severe form of dystrophinopathy, much like Becker muscular dystrophy. Eteplirsen's proposed mechanism of action is to bind to pre-mRNA needed to make a particular muscle protein, dystrophin, and rearrange the splicing of the RNA so that more dystrophin is made. By increasing the quantity of an abnormal, but potentially functional, dystrophin protein, the objective is to slow or prevent the progression of DMD.[10][12]
Nature and sequence of oligo and target
Eteplirsen is a morpholino phosphorodiamidate antisense oligomer.
CTCCAACATCAAGGAAGATGGCATTTCTAG (sequence source: US FDA ETEPLIRSEN BRIEFING DOCUMENT NDA 206488[10]),
30-mer,
20% G,
43% CG,
Predicted Tm: 88.9 °C at 10 μM oligo.
Oligo complement CTAGAAATGCCATCTTCCTTGATGTTGGAG
DMD-001 Exon 51, ENST00000357033.8 in Ensembl.org, RNA target site marked. Given that the target site is within an exon, this is likely blocking binding of an exonic splice enhancer protein and so altering splicing by interfering with splice regulation. CTCCTACTCAGACTGTTACTCTGGTGACACAACCTGTGGTTACTAAGGAAACTGCCATCT CCAAA[CTAGAAATGCCATCTTCCTTGATGTTGGAG]GTACCTGCTCTGGCAGATTTCAACC GGGCTTGGACAGAACTTACCGACTGGCTTTCTCTGCTTGATCAAGTTATAAAATCACAGA GGGTGATGGTGGGTGACCTTGAGGATATCAACGAGATGATCATCAAGCAGAAG
Pharmacokinetics
Following single or multiple intravenous infusions, the majority of drug elimination occurred within 24 hours of intravenous administration. Elimination half-life of eteplirsen was 3 to 4 hours.[7]
History
New Drug Applications (NDA) for eteplirsen and a similar drug drisapersen were filed with the US Food and Drug Administration (FDA) in August 2015.[13] The Prescription Drug User Fee Act (PDUFA) goal dates for these were December 27, 2015 for drisapersen and February 26, 2016 for eteplirsen. Following FDA rejection of drisapersen, the agency announced a three-month time extension for its review of eteplirsen. The FDA panel decision was controversial because the FDA staff and the panel used a stricter standard of evidence than Sarepta and patient groups used. The FDA panel said that it was required by law to apply the standard of "substantial evidence" of effectiveness. This required randomized, controlled trials showing effectiveness of a meaningful clinical outcome, such as the ability to function in daily life. Sarepta and patient groups wanted to use the standard of historical controls, personal testimonies, and the presence of altered dystrophin in the body. On April 25, 2016, the Advisory Committee Panel voted against approval;.[14] However, in June 2016, FDA requested for additional data from Sarepta to confirm findings of dystrophin production by eteplirsen. Janet Woodcock, director of the FDA's Center for Drug Evaluation and Research, overruled the panel, and FDA Commissioner Robert Califf deferred to her decision. Eterplirsen received accelerated approval on September 19, 2016.[15]
The European Medicines Agency reviewed the molecule in 2018 and refused to approve it.[16]
Society and culture
The US list price of eteplirsen is $300,000 per year of treatment. The Institute for Clinical and Economic Review has found the drug not cost effective at the list price when the cost of 1 QALY was equal to $150,000.[6]
References
- Scoto, M; Finkel, R; Mercuri, E; Muntoni, F (August 2018). "Genetic therapies for inherited neuromuscular disorders". The Lancet. Child & Adolescent Health. 2 (8): 600–609. doi:10.1016/S2352-4642(18)30140-8. PMID 30119719.
- Lim, KR; Maruyama, R; Yokota, T (2017). "Eteplirsen in the treatment of Duchenne muscular dystrophy". Drug Des Devel Ther. 11: 533–545. doi:10.2147/DDDT.S97635. PMC 5338848. PMID 28280301.
Eteplirsen is applicable for approximately 14% of patients with DMDmutations
- "FDA grants accelerated approval to first drug for Duchenne muscular dystrophy". Press Announcements. U.S. Food & Drug Administration. September 19, 2016. Retrieved September 19, 2016.
- "Railroading at the FDA". Nature Biotechnology. 34 (11): 1078. November 2016. doi:10.1038/nbt.3733. PMID 27824847.
- Kounang, Nadia (4 October 2016). "The families that fought for controversial new drug". CNN. Retrieved 29 November 2016.
- "ICER sees current DMD therapies as too pricey, but notes data limitations". BioPharma Dive. Retrieved 2019-12-14.
- "Eteplirsen - prescribing information" (PDF). FDA. September 2016.
- Anthony, Karen; Feng, Lucy; Arechavala-Gomeza, Virginia; Guglieri, Michela; Straub, Volker; Bushby, Katherine; Cirak, Sebahattin; Morgan, Jennifer; Muntoni, Francesco (17 Oct 2012). "Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen". Hum Gene Ther Methods. 23 (5): 336–345. doi:10.1089/hgtb.2012.117. PMID 23075107.
- Moulton, HM; Moulton, JD (17 Feb 2010). "Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy". Biochim Biophys Acta. 1798 (12): 2296–2303. doi:10.1016/j.bbamem.2010.02.012. PMID 20170628.
- https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM497063.pdf
- Leppert, Brian J.; Kole, Ryszard (2012-07-26). "Targeting mRNA Splicing as a Potential Treatment for Duchenne Muscular Dystrophy". Discovery Medicine. 14 (74): 59–69. PMID 22846203.
- Mendell, Jerry; Rodino-Klapac, Louise R; Sahenk, Zarife; Roush, Kandice; Bird, Loren; Lowes, Linda P; Alfano, Lindsay; Gomez, Ann Maria; Lewis, Sarah; Kota, Janaiah; Malik, Vinod; Shontz, Kim; Walker, Christopher M; Flanigan, Kevin M; Kean, John R; Allen, Hugh D; Shilling, Chris; Melia, Kathleen R; Sazani, Peter; Saoud, Jay B; Kaye, Edward M; Kaye, Edward M. (2013). "Eteplirsen for the treatment of duchenne muscular dystrophy". Ann. Neurol. 74 (5): 637–647. doi:10.1002/ana.23982. PMID 23907995.
- "FDA Accepts Sarepta's NDA for Eteplirsen". Rare Disease Report. Archived from the original on 2015-08-28. Retrieved 2015-08-28.
- Pollack, Andrew (2016-04-25). "Advisers to F.D.A. Vote Against Duchenne Muscular Dystrophy Drug". The New York Times.
- Column: To appease a patient lobby, did the FDA approve a $300,000 drug that doesn't work? Michael Hiltzik, Los Angeles Times, October 28, 2016
- "Going Its Own Way, European Regulators Reject Sarepta's Exondys 51 for DMD". BioSpace. Retrieved 2019-12-14.