Sack–Barabas syndrome
Sack–Barabas syndrome is an older name for the medical condition Ehlers-Danlos syndrome, vascular type. It affects the body's blood vessels and organs, making them prone to rupture.
| Sack–Barabas syndrome | |
|---|---|
| Other names | vEDS[1] |
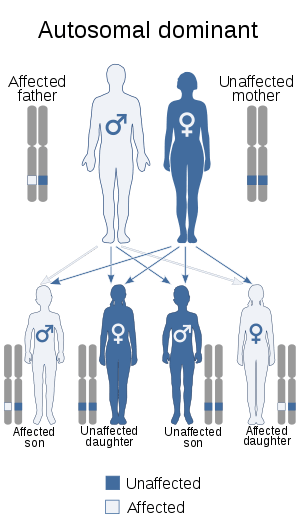 | |
| This condition is inherited in an autosomal dominant manner | |
Signs and symptoms
Patients with Sack–Barabas syndrome have thin, fragile skin, especially in the chest and abdomen, that bruises easily; hands and feet may have an aged appearance. Skin is soft but not overly stretchy.
Facial features are often distinctive, including protruding eyes, a thin nose and lips, sunken cheeks, and a small chin.
Other signs of the disorder include hypermobility of joints, tearing of tendons and muscles, painfully swollen veins in the legs, lung collapse, and slow wound healing following injury or surgery.
Infants with the condition may be born with hip dislocations and clubfeet.
Unpredictable ruptures of arteries and organs are serious complications of SBS. Ruptured arteries can cause internal bleeding, stroke, or shock, the most common cause of death in patients with this disorder.
Rupture of the intestine is seen in 25 to 30 percent of affected individuals and tearing of the uterus during pregnancy affects 2 to 3 percent of women. Although these symptoms are rare in childhood, more than 80 percent of patients experience severe complications by the age of 40. Teenage boys are at high risk for arterial rupture, often being fatal.
Causes
Sack–Barabas is caused by mutations in the COL3A1 gene.
- About half of all cases are inherited from a parent who has the condition. The condition is inherited in an autosomal dominant pattern, which means only one copy of the altered gene is necessary to cause the disorder.
- The other half of cases occurs in patients whose families have no history of the disorder. These sporadic cases are caused by new mutations in one copy of the COL3A1 gene.
The protein determined by the COL3A1 gene is used to assemble larger type III collagen molecules, found mostly in skin, blood vessels, and internal organs.
When the structure or production of type III collagen is altered by a mutation in the COL3A1 gene, collagen fibrils cannot be assembled properly in these tissues, and the symptoms of Ehlers-Danlos syndrome result.
Diagnosis
The tests to verify Sack–Barabas syndrome are biochemical samples such as collagen typing (performed on a skin biopsy sample) or collagen gene mutation testing. There is no cure for Ehlers-Danlos syndrome, so individual problems and symptoms must be evaluated and cared for accordingly.
Treatment
The key for managing Sack–Barabas syndrome is for the affected person to be aware of their disease. Close follow up and planning of interventions can significantly prolong and maintain the quality of life of a person with this disease.
Pregnant affected women must take special care due to the increased risk of premature death due to rupture of arteries, bowel or uterine rupture with a reported mortality rate of 50%.
Genetic counseling is recommended for prospective parents with a family history of Ehlers–Danlos syndrome. Affected parents should be aware of the type of Ehlers-Danlos syndrome they have and its mode of inheritance.
Epidemiology
Sack–Barabas syndrome is rare and has an estimated prevalence of 1 in 100,000 to 200,000.
The initial clinical manifestation of vascular problems in patients with SBS is early, about 25% have their first symptoms at age 20 and more than 80% of patients have had at least one complication by the age of 40.
The median survival for one study of SBS patients was only 48 years.
History
The condition was originally named for German physician Georg Sack, who described a single case in 1936,[2] and British surgeon A.P. Barabas, who described two cases in 1967.[3] Barabas recognized that the condition was a form of Ehlers-Danlos syndrome, a group of inherited disorders affecting connective tissue. This condition is now called Ehlers–Danlos syndrome, vascular type [formerly EDS type IV].
See also
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Vascular Ehlers Danlos syndrome". www.orpha.net. Retrieved 30 October 2019.
- Sack G (1936). "Status Dysvascularis, ein Fall von besonderer Zerreis-slichkeit der Blutgefässe". Deutsches Archiv für Klinische Medizin. 178: 663–669.
- Barabas A.P. (1967). "Heterogeneity of the Ehlers-Danlos Syndrome: Description of Three Clinical Types and a Hypothesis to Explain the Basic Defect(s)" (PDF). British Medical Journal. 2 (5552): 612–3. doi:10.1136/bmj.2.5552.612. PMC 1842124. PMID 6025600.
External links
| Classification | |
|---|---|
| External resources |
|