Discovery and development of triptans
Triptans is a word commonly used for a class of anti-migraine drugs that are selective 5-hydroxytryptamine/serotonin1B/1D (5-HT1B/1D) agonists.[1] Migraine is a complex disease which affects about 15% of the population and can be highly disabling.[2] Triptans have advantages over ergotamine and dihydroergotamine, such as selective pharmacology, well established safety record and evidence-based prescribing instructions. Triptans are therefore often preferred treatment in migraine.[1]
History
Search for a new anti-migraine drug started at Glaxo in 1972. Studies in the 1960s showed that vasoconstriction from 5-HT, ergotamine and noradrenaline could reduce migraine attacks. Research also showed that platelet 5-HT level is reduced during migraine. Because there are too many side-effects for 5-HT to be used as a drug, scientists started research on the receptors of 5-HT in order to discover and develop a more specific agonist for 5-HT receptors. Research on the 5-HT receptors and their effect led to discovery of several types and subtypes of 5-HT. AH24167 showed a vasodilation effect instead of vasoconstriction due to the agonist effect on another type of 5-HT receptors later assigned the name 5-HT7. AH25086 was the second compound developed and showed a vasoconstriction effect but was not released as a drug due to low per oral bioavailability. Continued research led to the discovery of the first triptan drug, sumatriptan, that had both vasoconstriction effect, as well as better oral bioavailability. Sumatriptan was first launched in the Netherlands in 1991 and became available in the United States during 1993.[3]
Mechanism
Triptans are specific and selective agonists for the 5-HT1 receptors. Sumatriptan[4] binds to 5-HT1D receptors, zolmitriptan,[5] rizatriptan,[6] naratriptan,[7] almotriptan,[8] and frovatriptan[9] binds to 5-HT1B/1D and eletriptan[10] binds to 5-HT1B/1D/1F receptors. Triptans are believed to exert their effects through vasoconstriction, leading to reduced carotid arterial circulation without affecting cerebral blood flow, peripheral neuronal inhibition, or inhibition of transmission through second order neurons of the trigeminocervical complex. [1]
Receptors
5-HT receptors are all G-protein coupled receptors (GPCR) except for 5-HT3 which is a ligand gated ion channel. The receptors that have been found to be involved in migraine are 5-HT1B, 5-HT1D and 5-HT1F receptors. 5-HT1B are found in meningeal arteries, agonism of 5-HT1B causes vasoconstriction in cranial nerves. The 5-HT1D receptors are located primarily in the trigeminal nerve in the central nervous system (CNS). They are also found in vascular smooth muscles, mediating contraction. Agonism of 5-HT1D receptors subdues the release of inflammatory inducing nerve stimulants. The amino acids contributing to the binding of ligands to the receptor are aspartic acid (Asp), phenylalanine (Phe), serine (Ser), threonine (Thr), tryptophan (Trp) and tyrosine (Tyr). It has been shown that both 5-HT1B and 5-HT1D receptors in humans have a very similar amino acids structures which demonstrates the similarities in binding properties.[11][12][13]
Design
All triptans have an indole structure identical to the neurotransmitter 5-HT. Classic triptan structure contain side chain on the indole ring, and a basic nitrogen in a similar distance from the indole structure. The main structural difference of the triptans is the position of the sulfonamide and the side chain attached to it (see figure 1 and table 1). Rizatriptan and zolmitriptan have instead of a sulfonamide a triazole and 2-oxazolidone respectively. Another exception to the classic structure is seen on eletriptan where the nitrogen-alkyl chain connected to the indole ring is replaced with a dimethyl-pyrrolidine, and in naratriptan where the nitrogen-alkyl chain is replaced with a 1-methyl-piperidine ring.
One of the frovatriptan side chains forms an additional ring with the indole, resulting in a carbazole ring system.
Structures of the triptans
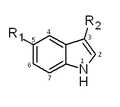 Fig 1. Indole structure of triptans
Fig 1. Indole structure of triptans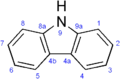 Fig 1a. Carbazole structure of frovatriptan
Fig 1a. Carbazole structure of frovatriptan
| Triptan | R1 | R2 | Triptan | R1 | R2 |
|---|---|---|---|---|---|
| Sumatriptan |  |
 |
Eletriptan | 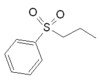 |
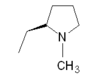 |
| Rizatriptan | 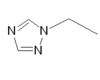 |
|
Naratriptan | 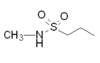 |
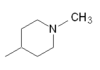 |
| Almotriptan | 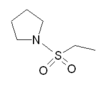 |
|
Frovatriptan |  |
 |
| Zolmitriptan | 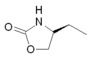 |
|
– | – | – |
The 5-HT1B/D pharmacophore
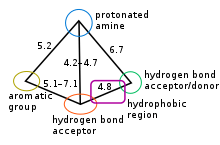
5-HT1B and 5-HT1D receptors are considered very similar, they share amino acid homology and their ligands expose similar binding properties thus they have similar pharmacophore. The pharmacophore model for these receptors ligands is qualitative and defines the relative positions of important groups. It is defined with following five main features: an aromatic group (usually the indole), protonated amine (a donor of hydrogen bond), acceptor of hydrogen bond, additional hydrogen bond site (both donor and acceptor) and hydrophobic region located between both hydrogen bond sites, see figure 2.[11][14] The main binding points were concluded to be the protonated amine and the hydrogen bond site. It was observed that the double bond region in the indole was necessary for the agonism in this series of compounds. Figure 3 shows how different drugs fit the pharmacophore, with an C and N linked analogues of 5-HT1D agonist. The marked sites on the figure are responsible for the affinity.[14][15] The pharmacophore can be characterized as amphipathic, that means that the structure has both hydrophobic and hydrophilic groups.[16]
Relevant structural features of triptans and binding to the receptor
Triptan structures were designed from the structure of 5-HT to attain affinity to 5-HT receptors, hence the identical indole structure. The hydroxyl group (-OH) on the hexane of the indole core and the alkyl-amine side chain on position C3 on 5-HT have been replaced with other compounds, such as sulfonamides or azol-ring structured derivatives and different amine-alkyl side chains. An electro-negative group can form a hydrogen bond with Thr in the pocket of the receptor. Sulfonamide derivatives attached to the hexane ring of the indole structure have electro-negative properties, as well as the triazole and 2-oxazolidone on rizatriptan and zolmitriptan respectively. This can increase binding ability of the compound and the efficacy, especially with the 5-HT1D receptor.[11]
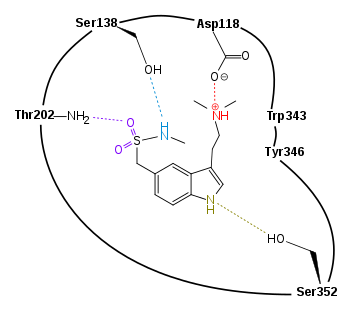
A schematic drawing of the binding of sumatriptan to 5-HT1D receptor can be seen in figure 4. One study[11] showed that sumatriptan fits better in the binding site of the receptor when the side chain with the protonated nitrogen atom is folded back over the indole structure. This alignment contributes to the hydrogen bonding between the nitrogen in the sulfonamine and the Ser138 in the binding site. It is also favorable to the formation of the hydrogen bond between the oxygen of the sulfonamine and Thr202. Other binding in the pocket of the binding site occurs with the nitrogen atom in the pentene ring of the indole structure of the triptan and the amino acid Ser352. This energetically favorable position of the agonist makes it possible for additional binding of the ligand to other Ser in the binding site, along with additional anchoring between Phe in the pocket of the binding site and the indole of the agonist. The binding of Phe and the triptan is caused by π stacking interactions of the indole and amino acid and an additional effect on this interaction is because of dispersive effect of amino acid leucine (Leu; not shown in figure 4). The amino acids Trp343 and Tyr346 both have electron rich π-systems in their aromatic structures. With their position in the binding site they create a sort of aromatic cage around the protonated nitrogen atom of the side chain on position C3 on the triptans (this nitrogen atom is protonated at physiological condition), and thereby stabilizes the ion bond the nitrogen atom has formed with a carboxylate on aspartic acid. Side chains of the surrounding amino acids can have an effect on the binding of the nitrogen atom, mainly three Phe can affect the methyl groups bound to the nitrogen atom (not shown in figure 4).[11][12][13]
Eletriptan has higher affinity for the receptor, which is probably a result of the bulky substituents of the structure. The amine is protonated at physiological pH condition, triggering better uptake.[16][17] The uptake rate of the agonist is different depending on whether the amine in R2 is primary, secondary or tertiary but the latter seem to give the best results. For the R1 substituent an electron rich sulfonamide groups and amide group has shown the best results in receptor binding and activity.[16] It has been observed that a relationship is between absorption and molecular size hence larger hydrophilic molecules tended to have poor absorption. A small R1 substituent is necessary to maintain the rapid oral bioavailability of triptans.[15]
By placing an electron-withdrawing group or large group on position C2 on the indole structure the 5-HT agonist is conversed into an antagonist. This is thought to be because the indole ring is unable to occupy the aromatic part of the binding site.[12]
Triptan drugs
Properties of formulations
Sumatriptan was the pioneer drug in this class. The second generation's triptans such as zolmitriptan, naratriptan, rizatriptan, almotriptan, eletriptan and frovatriptan soon became available.[18] Different triptans are available in different formulations and in different strengths (see table 2). They have been formulated as subcutaneous injections, oral tablets, orally disintegrating tablets, nasal spray and as rectal suppositories. Delivery system of the triptans may play an important role in the onset of action. The selection of anti-migraine drug for patients depends on their symptoms. The first selective 5-HT1B/1D agonist, sumatriptan, was first synthesized as a subcutaneous injection, then as an oral tablets and more recently as a nasal spray, it is also available in some countries as suppositories. The subcutaneous injection is the fastest way to stop a rapidly progressing migraine attack. The sumatriptan nasal spray provides faster onset of action than the tablets but it produces a similar headache response at 2 hours. Some patients prefer the nasal spray as it works more rapidly than the tablets and does not have as many adverse effects as the subcutaneous injection. Nasal spray is although not suitable for all patients, because some patients experience bad taste and lack of consistency of response. Zolmitriptan was developed with the strategy to create a more lipophilic compound, with faster absorption and better ability to cross the blood brain barrier than sumatriptan. It is available as tablets, orally disintegrating tablets and as nasal spray in some countries. Rizatriptan is available as tablets and orally disintegrating tablets but naratriptan, almotriptan, eletriptan and frovatriptan are only available in tablets, for now.[19]
| Generic | Formulations[19] | Doses (mg)[19] | Maximum daily dose (mg)[19] |
Onset of action (min)[20] |
Duration
of action[20] |
Affinity (pKI in nM) | Metabolism[21] | Excretion[20] |
|---|---|---|---|---|---|---|---|---|
| Sumatriptan |
Tablets |
25, 50, 100 |
200 |
Short | 7.9–8.5 | MAO-A |
Urine (57%), | |
| Zolmitriptan |
Tablets |
2.5, 5 |
10 |
45 |
Short | 9.2 |
CYP1A2 |
Urine (65%), |
| Naratriptan | Tablets | 1, 2.5 |
5 |
60–180 | Long | 8.3 |
CYPa |
Urine |
| Rizatriptan |
Tablets |
5, 10 |
30 |
30–120 |
Short | 7.7 | MAO-A | Urine |
| Almotriptan | Tablets | 6.25, 12.5 | 25 | 60–180 | Short | 7.8 |
MAO-A |
Urine (40%), |
| Eletriptan | Tablets | 20, 40 | 80 | <60[23] | – | 8.9 | CYP3A4 | – |
| Frovatriptan | Tablets | 2.5 | 7.5 | 60–120 | Long | 8.4 | CYP1A2 | Urine (40%) |
a Specific enzyme not yet reported.
The U.S. Food and Drug Administration (FDA) approved a new drug April 15, 2008, which is a combination of sumatriptan 85 mg and naproxen 500 mg (NSAID).[24] Triptans and NSAIDs work on distinct mechanism involved in migraine and therefore may offer improved treatment when administrated together.[25]
Pharmacokinetics
Pharmacokinetic properties (see table 3) are important when new drugs are developed.[26]
Patients seek rapid onset of action to relief the headache. Relatively short tmax, good bioavailability and lipophilicity are pharmacokinetic properties that have been associated with rapid onset of action. It has been speculated that good ability to cross the blood brain barrier and relatively long terminal elimination half-life may result in a lower incidence of headache recurrence. Sumatriptan and rizatriptan undergo first pass hepatic metabolism and result in lower bioavailability.[18]
| Generic | Bioavailability (%)[26] | Lipophilicity[19] | Protein binding (%)[20] |
t1/2 (h)[26] | tmax (h)[22] | ClR (mL min-1)[27] |
Log DpH7.4[28] | VD[20] |
|---|---|---|---|---|---|---|---|---|
| Sumatriptan | 14 | Low | 10–21 | 2–2.5 | 2–2.5 | 260 | -1.5 | 2.4–3.3 L/kg |
| Zolmitriptan | 40 | Moderate | 25 | 3 | 2 | 193 | -1.0 | 7.0 L/kg |
| Naratriptan | 63(M) / 74(F) | High | 28–31 | 5–6 | 2–3 | 220 | -0.2 | 2.4 L/kg |
| Rizatriptan | 47 | Moderate | 14 | 2–2.5 | 1.3 | 414 | -0.7 | 140(M) / 110(F) L |
| Almotriptan | 69 | – | 35 | 3.6 | 1.4–3.8 | – | -2.1 | 180–200 L |
| Eletriptan | 50 | High | 85[10] | 4–5 | 1–2 | 597 | 0.5 | 138 L[10] |
| Frovatriptan | 24(M) / 30(F) | Low | 20–30 | 25[18][26] | 2–4 | 216(M) / 132(F)[9] | -1.0[29] | 4.2(M) / 3.0(F) L/kg |
t1/2 = Elimination half-life;
tmax = Time to reach peak plasma drug concentration;
ClR = Renal Clearance;
LogDpH7.4 = Measure of lipophilicity at pH 7.4. Increasing number indicate greater solubility;
VD = Volume of distribution
M = Male; F = Female
Future research
Most triptans were developed and introduced in the 1990s. Further studies have not shown much promise regarding the development of new triptans with better duration of action, efficacy and safety profile. Therefore, it is unlikely that further variations will be developed and new anti-migraine drugs are likely to have another mechanism of action.[29]
References
- Ferrari, M. D.; Goadsby, P.J.; Roon, K. I.; Lipton, R.B. (2002), "Triptans (serotonin, 5-HT1B/1D agonists) in migraine: detailed results and methods of a meta-analysis of 53 trials", Cephalalgia, 22 (8): 633–658, doi:10.1046/j.1468-2982.2002.00404.x, PMID 12383060, archived from the original on 2012-12-17
- Goadsby, Peter J. (2006), "Recent advances in understanding migraine mechanism, molecules and therapeutics", Trends in Molecular Medicine, 13 (1): 39–44, doi:10.1016/j.molmed.2006.11.005, PMID 17141570
- Humphrey, Patrick P.A. (2007), "The Discovery of a New Drug Class for the Acute Treatment of Migraine", Headache, 47 [Suppl 1]: 10–19, doi:10.1111/j.1526-4610.2007.00672.x, PMID 17425704
- "Imigran Tablets 50mg Imigran Tablets 100mg". Retrieved 2008-11-09.
- "Zomig Tablets 2.5mg". Retrieved 2008-11-09.
- "Maxalt 5mg, 10mg Tablets, Maxalt Melt 10mg Oral Lypophilisates". Archived from the original on 2008-05-02. Retrieved 2008-11-09.
- "Naramig Tablets 2.5mg". Retrieved 2008-11-09.
- "Axert". Retrieved 2008-11-09.
- "Migard". Retrieved 2008-11-09.
- "Relpax – 20 mg and 40 mg". Retrieved 2008-11-09.
- Bremner, DH; Ringan, NS; Wishart, G (1997), "Modeling of the agonist binding site of serotonin human 5-HT1A, 5-HTDα and 5-HTDβ receptors", European Journal of Medicinal Chemistry, 32 (1): 59–69, doi:10.1016/S0223-5234(97)84362-0
- Bojarski, Andrzej J. (2006), "Pharmacophore Models for Metabotropic 5-HT Receptor Ligands", Current Topics in Medicinal Chemistry, 6 (18): 2005–2026, doi:10.2174/156802606778522186, PMID 17017971
- Terzioglu, Nalan; Höltje, Hans-Dieter (2005), "Receptor-based 3D QSAR Analysis of Serotonin 5-HT1D receptor agonists", Collection of Czechoslovak Chemical Communications, 70 (9): 1482–1492, doi:10.1135/cccc20051482
- Buckingham, Janet; Glen, Robert C.; Hill, Alan P.; Hyde, Richard M.; Martin, Graeme R.; Robertson, Alan D.; Woollard, Patrick M. (1995). "Computer-Aided Design and Synthesis of 5-Substituted Tryptamines and Their Pharmacology at the 5-HT1D Receptor: Discovery of Compounds with Potential Anti-Migraine Properties". Journal of Medicinal Chemistry. 38 (18): 3566–3580. doi:10.1021/jm00018a016.
- Jandu, K. S.; Barrett, V.; Brockwell, M.; Cambridge, D.; Farrant, D. R.; Foster, C.; Selwood, D. L. (2001). "Discovery of 4-[3-(trans-3-Dimethylaminocyclobutyl)-1H-indol-5-ylmethyl]- (4S)-oxazolidin-2-one (4991W93), a 5HT1B/1D Receptor Partial Agonist and a Potent Inhibitor of Electrically Induced Plasma Extravasation". Journal of Medicinal Chemistry. 44 (5): 681–693. doi:10.1021/jm000956k.
- Cheng, Ziqiang; Liu, Houfu; Yu, Na; Wang, Fei; An, Gang; Xu, Yan; Ayrton, Andrew (2012). "Hydrophilic anti-migraine triptans are substrates for OATP1A2, a transporter expressed at human blood-brain barrier". Xenobiotica. 42 (9): 880–890. doi:10.3109/00498254.2012.675455. PMID 22509823.
- Street, Leslie J.; Baker, Raymond; Castro, Jose L.; Chambers, Mark S.; Guiblin, Alexander R.; Hobbs, Sarah C.; Beer, Margaret S. (1993). "Synthesis and serotonergic activity of 5-(oxadiazolyl)tryptamines: potent agonists for 5-HT1D receptors". Journal of Medicinal Chemistry. 36 (11): 1529–1538. doi:10.1021/jm00063a003. PMID 8496922.
- Mathew, Ninan T.; Loder, Elizabeth W. (2005), "Evaluating the triptans", The American Journal of Medicine, 118 (12): 28–35, doi:10.1016/j.amjmed.2005.09.014, PMID 16356805
- Bigal, Marcelo E.; Bordini, Carlos A.; Antoniazzi, Ana L.; Speciali, José G (2003), "The triptan formulations, A critical evaluation" (PDF), Arquivos de Neuro-Psiquiatria, 61 (2A): 313–320, doi:10.1590/s0004-282x2003000200032, PMID 12806521
- "Drug class review: Oral 5HT1 Receptor agonists" (PDF). U.S. Department of Veterans Affairs. Archived from the original (PDF) on 2009-01-14. Retrieved 2008-11-03.
- Armsterong, Scott C.; Cozza, Kelly L. (2002), "Triptans", Psychosomatics, 43 (6): 502–504, doi:10.1176/appi.psy.43.6.502, PMID 12444236, archived from the original on 2003-07-12
- Rapoport, Alan M.; Tepper, Stewart J.; Sheftell, Fred D.; Kung, Edna; Bigal, Marcelo E. (2006), "Which triptan for which patient?", Neurological Sciences, 27: 123–129, doi:10.1007/s10072-006-0586-y, PMID 16688615
- Färkkilä, M.; Dalhlöf, C.; Stovner, L.J.; Bruggen, J.P ter; Rasmussen, S.; Muirhead, N.; Sikes, C.; Sikes, C (2003), "Eletriptan for the treatment of migrain in patients with previous poor response or toletance to oral sumatriptan", Cephalalgia, 23 (6): 463–471, doi:10.1046/j.1468-2982.2003.00554.x, PMID 12807526, archived from the original on 2012-12-17
- "press release - Treximet (sumatriptan and naproxen sodium) tablets approved by FDA for acute treatment of migraine". GlaxoSmithKline. Archived from the original on 2008-12-04. Retrieved 2008-11-09.
- Smith, Timothy R.; Sunshine, Abraham; Stark, Stuart R.; Littlefield, Diane E.; Spruill, Susan E.; Alexander, W. James (2005), "Sumatriptan and Naproxen Sodium for the Acute Treatment of Migraine", Headache, 45 (8): 983–991, doi:10.1111/j.1526-4610.2005.05178.x, PMID 16109111, archived from the original on 2012-12-18
- Jhee, Stanford S.; Shiovitz, Thomas; Crawford, AAron W.; Cutler, Neal R. (2001), "Pharmacokinetics and pharmacodynamics of the triptan antimigraine agents", Clinical Pharmacokinetics, 40 (3): 189–205, doi:10.2165/00003088-200140030-00004, PMID 11327198
- Saxena, Pramod R.; Tfelt-Hansen, Peer (2001), "Success and failure of triptans", The Journal of Headache and Pain, 2: 3–11, doi:10.1007/s101940170040
- Pascual, Julio; Muñoz, Pedro (2005), "Correlation between lipophilicity and triptan outcomes", Headache, 45 (1): 3–6, doi:10.1111/j.1526-4610.2005.05003.x, PMID 15663606
- Lambert, Geoffrey A. (2005), "Preclinical Neuropharmacology of Naratriptan", CNS Drug Reviews, 11 (3): 289–316, doi:10.1111/j.1527-3458.2005.tb00048.x, PMC 6741765, PMID 16389295