Discovery and development of 5α-reductase inhibitors
This article is about the discovery and development of 5α-reductase inhibitors (5-ARIs), also known as dihydrotestosterone (DHT) blockers.
Development of 5α-reductase inhibitors
These are two types of 5-ARIs, categorized as steroidal and nonsteroidal 5-ARIs.[1]
Steroidal 5α-reductase inhibitors
Steroid 5α-reductase is a membrane-associated enzyme in an oxidoreductase family and has an important role in biological actions towards steroid metabolism. If the steroid 5α-reductase is overexpressed it causes overproduction of DHT that can lead to androgenic disorders in humans.[2]
The 5α-reductase isozymes possess a similar steroidal catalytic site.[2] The only available information about the 5α-reductase isozymes is their primary sequence estimated from c-DNAs and that affects the design of the novel inhibitors. The crystal structure of the 5α-reductase isozymes is not known because the nature of the 5α-reductase enzyme is so unstable during purification. The first 5-ARIs were designed by modifying the structure of natural substrates, including the substitution of one carbon atom of the rings of the steroids by a heteroatom such as nitrogen thereby forming azasteroids.[1] The receptor is known to consist of two hydrogen bond donors, where the C3 and 17β-side chain of the ligands connect, as well as three hydrophobic groups distributed over the steroidal structure. The best receptor inhibitors comply with these factors.[3] Azasteroids are a type of steroid derivatives which have nitrogen atoms replaced at various positions for one of the carbon atoms in the steroid ring system. Two 4-azasteroids, finasteride and dutasteride are marketed as 5-ARIs.[1] Finasteride (Proscar or Propecia) was the first steroidal 5α-reductase inhibitor approved by the U.S. Food and Drug Administration (USFDA). It inhibits the function of two of the isoenzymes (type II and III).[4] In human it decreases the prostatic DHT level by 70–90% and reduces the prostatic size.[1]
Dutasteride (Avodart) was the second steroidal 5α-reductase approved after finasteride. It is a competitive inhibitor of all three 5α-reductase isoenzymes[4] and it inhibits types 1 and 2 better than finasteride, leading to it causing further reduction in DHT, with >90% recuded DHT levels following 1 year of oral administration.[1]
Epristeride is the third marketed steroidal 5-ARI. It is a noncompetitive, specific inhibitor. It potency is not as significant as finasteride or dutasteride and thus it is only marketed in China.[5]
Nonsteroidal 5α-reductase inhibitors
Various pharmaceutical and academic groups have conducted the synthesis of nonsteroidal compounds that inhibit human 5α-reductases due to the unwanted hormonal side effects of steroidal compounds. Nonsteroidal inhibitors can be categorized due to their structure. Many have been obtained from azasteroid inhibitors by taking away one or more rings from the steroid structure.
Four main categories of nonsteroidal 5-ARIs have been described:
- Benzo(c)quinolizinones
- Benzo(f)quinolonone
- Piperidones
- Carboxylic acids
Nonsteroidal inhibitors are thought to act as competitive inhibitors on the 5α-reductase isozymes, except for epristeride analogues (carboxylic acids), which are noncompetitive inhibitors.[6]
Bexlosteride falls into the category of benzo(f)quinolonones, and is probably the derivative that has come closest to being marketed. It functions as a 5-ARI1 inhibitor which inhibits testosterone stimulated LNCaP cell growth but without testosterone the compound shows no effect and was therefore never marketed.[7]
Structure–activity relationships
4-Azasteroids
Many steroidal 5-ARIs have been researched but only 3 are marketed. Two of them are 4-azasteroids and will be covered here. As mentioned above, the third one, epristeride is only marketed in China and will not be covered here. The basic SAR of 4-azasteroids is shown below. For competitive inhibiting functions there are two functions considered crucial, 4-en-3-one function and a lipophilic 17β-side chain with one or more oxygen atoms. The main problems for 4-azasteroids is the rapid conversion into inactive 4,5-dihydro form, which is done by the enzyme.[1]

Finasteride is considered similar to the transition state of reduced testosterone and is thus a slow-offset, irreversible inhibitor. The similarity to the transition state is a formation of an enzyme-NADP-dihydrofinasteride adduct by rearrangement on the A-ring of the compound.[8]
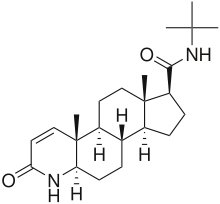
Finasteride mainly inhibits the 5α-R2 (IC50=69 nM) and 5α-R3 (IC50=17.4 nM) with little inhibition of 5α-R1 (IC50=360 nM). As mentioned above, finasteride reduces prostatic DHT levels on a 70-90% range but the detailed reduction of DHT is 70.8% and 85% of intraprostatic DHT.[6]
Dutasteride, however, is a so-called dual inhibitor with both 5α-R1 and 5α-R2 inhibition. IC50 for 5α-R1 is 7 nM but 6 nM for 5α-R2. As mentioned above, it reduces DHT > 90% overall, or precisely 94.7% and for intraprostatic DHT the reduction is 97-99%. Dutasteride has also been found to inhibit 5α-R3, in vitro, with IC50=0.33 nM.[6] The 2,5-difluorophenyl side chain on the D-ring of the compound shows significant lipophilic features and as increased lipophilicity enhances the potency of the compounds binding at pocket site, its potency is much greater than of finasteride.[8]

Finasteride is an unsaturated analogue of another 4-azasteroid, or 4-MA. 4-MA is known to have dual inhibiting features with good inhibition on 5α-R1 (IC50=1.7 nM) and 5α-R2 (IC50=1.9 nM).[6] However, 4-MA was never marketed as it showed hepatotoxicity. There is no detailed data about the cause of hepatotoxicity in 4-MA regarding SAR, but a conclusion may be drawn that the R2 group is the cause as there are other 4-azasteroid compounds containing the same R1 group as 4-MA, or CH3, without showing hepatotoxicity.[1]
Nonsteroidal
The common factor in nonsteroidal 5-ARI discovery is that the first compounds were all selective inhibitors to 5α-reductase type 1 only, but were then developed in order to get dual inhibition on both type 1 and 2, since inhibition of the type 2 isozyme is a more important factor in treating the disease of BPH.[6][8]
Benzo(c)quinolizinones are tricyclic derivatives of 10-azasteroids. The D-ring has been removed and the C-ring substituted for an aromatic one.[6] The first compounds developed were selective 5-alpha reductase type 1 inhibitors, but the most potent one inhibits both type 1 and 2. The fluorine atom is an important part of the structure.[8]
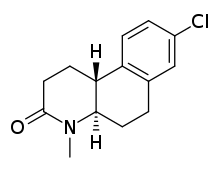
Benzo(f)quinolonone are also tricyclic compounds, but derivatives of the 4-azasteroid structure. The compounds that have been designed can be divided into two categories, hexahydro derivatives and octahydro derivatives. The octahydro derivatives have been proven to be more potent.[8] Compound LY 191704, later named bexlosteride, is the most potent octahydro derivative designed. It is a selective inhibitor to the type 1 isozyme, especially because of the chlorine atom and the amino-methyl group.[6]
Piperidones are also 4-azasteroid derivatives but both B- and D-ring have been removed. The original compounds designed were type 1 selective, especially the ones containing a chlorine atom connected to the aromatic ring. By inserting a styryl group to the piperidones type 2 inhibitory activity increased.[8]
Nonsteroidal carboxylic acids are tricyclic compounds designed to resemble steroidal carboxylic acids such as episteride. As with the other nonsteroidal inhibitors, they have been designed by removing steroid ring systems.[8] As with the piperidones, addition of a styryl group provides good dual inhibition on isozyme 1 and 2, but the nonsteroidal carboxylic acids are mostly type 1 selective.[6]

Natural products

Saw palmetto extract
The European Medicines Agency (EMA) has concluded that the extract of the natural product Saw palmetto can be used to treat symptoms of benign prostatic hyperplasia (BPH) as research has shown its 5-ARI effects.[9] An extract of Serenoa repens, also known as saw palmetto extract, is a 5-ARI that is sold as an over-the-counter dietary supplement. It is also used under the brand name Permixon in Europe as a pharmaceutical drug for the treatment of benign prostatic hyperplasia.
References
- Aggarwal, Saurabh; Thareja, Suresh; Verma, Abhilasha; Bhardwaj, Tilak Raj; Kumar, Manoj (2010). "An overview on 5α-reductase inhibitors". Steroids. 75 (2): 109–153. doi:10.1016/j.steroids.2009.10.005. PMID 19879888.
- Karnsomwan, Wiranpat; Rungrotmongkol, Thanyada; De-Eknamkul, Wanchai; Chamni, Supakarn (2016-06-01). "In silico structural prediction of human steroid 5α-reductase type II". Medicinal Chemistry Research. 25 (6): 1049–1056. doi:10.1007/s00044-016-1541-y. ISSN 1054-2523.
- Chen, Grace Shiahuy; Chang, Chih-Shiang; Kan, Wai Ming; Chang, Chih-Long; Wang, K. C.; Chern, Ji-Wang (2001-11-01). "Novel Lead Generation through Hypothetical Pharmacophore Three-Dimensional Database Searching: Discovery of Isoflavonoids as Nonsteroidal Inhibitors of Rat 5α-Reductase". Journal of Medicinal Chemistry. 44 (23): 3759–3763. doi:10.1021/jm010433s. ISSN 0022-2623.
- Yamana, Kazutoshi; Labrie, Fernand; Luu-The, Van (2010-08-01). "Human type 3 5α-reductase is expressed in peripheral tissues at higher levels than types 1 and 2 and its activity is potently inhibited by finasteride and dutasteride". Hormone Molecular Biology and Clinical Investigation. 2 (3): 293–9. doi:10.1515/hmbci.2010.035. ISSN 1868-1891. PMID 25961201.
- Borchardt, Ronald T.; et al. (2006). Integration of Pharmaceutical Discovery and Development. Google Books: Kluwer Academic Publishers. p. 398. ISBN 978-0-306-47384-5.
- Azzouni F, Godoy A, Li Y, Mohler J, et al. (2012). "The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases". Adv. Urol. 2012: 530121. doi:10.1155/2012/530121. PMC 3253436. PMID 22235201.
- Chang, Chawnshang (2005). Prostate Cancer: Basic Mechanisms and Therapeutic Approaches. Singapore: World Scientific Publishing. p. 250. ISBN 978-981-256-067-4.
- Kulig, Katarzyna., Malawska, Barbara (2006). "Trends in the Development of New Drugs for Treatment of Benign Hyperplasia". Current Medicinal Chemistry. 13: 3395–2416. doi:10.2174/092986706779010315.
- "Herbal medicine: Summary for the Public. Saw Palmetto Fruit" (PDF). www.ema.europa.eu. 5 April 2016. Retrieved 28 September 2017.