Carnitine palmitoyltransferase I deficiency
Carnitine palmitoyltransferase I deficiency is a rare metabolic disorder that prevents the body from converting certain fats called long-chain fatty acids into energy, particularly during periods without food.[1] It is caused by a mutation in CPT1A on chromosome 11.
| Carnitine palmitoyltransferase I deficiency | |
|---|---|
| Other names | CPT-I, CPT1 |
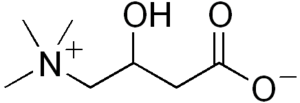 | |
| Carnitine | |
Carnitine, a natural substance acquired mostly through the diet, is used by cells to process fats and produce energy. People with this disorder have a faulty enzyme, carnitine palmitoyltransferase I, that prevents these long-chain fatty acids from being transported into the mitochondria to be broken down.
Symptoms
Signs and symptoms of this disorder include low levels of ketones (hypoketosis) and low blood sugar (hypoglycemia). Together these signs are called hypoketotic hypoglycemia. People with this disorder typically also have an enlarged liver (hepatomegaly), muscle weakness, and elevated levels of carnitine in the blood.[2]
Genetics
Mutations in the CPT1A gene cause carnitine palmitoyltransferase I deficiency by producing a defective version of an enzyme called carnitine palmitoyltransferase I. Without this enzyme, long-chain fatty acids from food and fats stored in the body cannot be transported into mitochondria to be broken down and processed. As a result, excessive levels of long-chain fatty acids may more rapidly build up in tissues, damaging the liver, heart and/or brain.
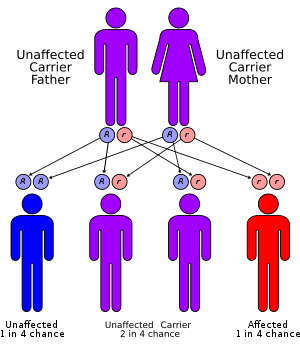
This condition has an autosomal recessive inheritance pattern, which means the defective gene must be inherited from both parents. The parents of a child with an autosomal recessive disorder are carriers of one copy of the defective gene, but are usually not affected by the disorder.
The prevalence of mutations associated with this condition reach 68% to 81% in certain arctic coastal populations, suggesting that the condition had some adaptive value in those habitats at some time.[3][4] Fewer than 50 people have been identified with this condition.[5]
Diagnosis
Genetic testing is necessary for diagnosis.
Differential diagnosis
This condition is sometimes mistaken for fatty acid and ketogenesis disorders such as Medium-chain acyl-coenzyme A dehydrogenase deficiency (MCAD), other long-chain fatty acid oxidation disorders such as Carnitine palmitoyltransferase II deficiency (CPT-II) and Reye syndrome.[6]
Treatment
Treatment for this condition is supportive. There is no cure. Affected people should eat a high-carbohydrate, low-fat diet and avoid fasting.[7]
References
- Bennett, Michael J.; Santani, Avni B. (1993-01-01). "Carnitine Palmitoyltransferase 1A Deficiency". In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Ledbetter, Nikki; Mefford, Heather C. (eds.). GeneReviews. Seattle (WA): University of Washington, Seattle. PMID 20301700.update 2010
- "Carnitine palmitoyl transferase 1 deficiency | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov.
- Clemente, Florian; al., et. (2014-11-06). "A Selective Sweep on a Deleterious Mutation in CPT1A in Arctic Populations". The American Journal of Human Genetics. AJHG. 95 (5): 584–589. doi:10.1016/j.ajhg.2014.09.016. PMC 4225582. PMID 25449608. Retrieved 2015-02-22.
- Greenberg, Cheryl; al., et. (2009-04-09). "The paradox of the carnitine palmitoyltransferase type Ia P479L variant in Canadian Aboriginal populations". Molecular Genetics and Metabolism. 96 (4): 201–7. doi:10.1016/j.ymgme.2008.12.018. PMID 19217814.
- Reference, Genetics Home. "CPT I deficiency". Genetics Home Reference.
- Bennett, Michael; Stanley, Charles (2011-03-01). "Carnitine palmitoyl transferase 1A deficiency". Orphanet. Retrieved 2014-12-04.
- Bennett, Michael J.; Santani, Avni B. (1993). "Carnitine Palmitoyltransferase 1A Deficiency". GeneReviews®. University of Washington, Seattle.
External links
| Classification | |
|---|---|
| External resources |
This article incorporates public domain text from The U.S. National Library of Medicine