Camptothecin
Camptothecin (CPT) is a topoisomerase inhibitor. It was discovered in 1966 by M. E. Wall and M. C. Wani in systematic screening of natural products for anticancer drugs. It was isolated from the bark and stem of Camptotheca acuminata (Camptotheca, Happy tree), a tree native to China used as a cancer treatment in Traditional Chinese Medicine.[1] CPT showed remarkable anticancer activity in preliminary clinical trials. However, it has low solubility, so synthetic and medicinal chemists have developed numerous syntheses of camptothecin[2][3][4] and various derivatives to increase the benefits of the chemical, with good results. Two CPT analogues have been approved and are used in cancer chemotherapy[5] today, topotecan and irinotecan.[6][7]
 | |
| Clinical data | |
|---|---|
| ATC code |
|
| Identifiers | |
IUPAC name
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.113.172 |
| Chemical and physical data | |
| Formula | C20H16N2O4 |
| Molar mass | 348.352 g/mol g·mol−1 |
| 3D model (JSmol) | |
| Melting point | 275 to 277 °C (527 to 531 °F) |
SMILES
| |
InChI
| |
| (verify) | |
Structures
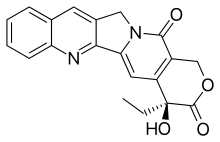
CPT has a planar pentacyclic ring structure, that includes a pyrrolo[3,4-β]-quinoline moiety (rings A, B and C), conjugated pyridone moiety (ring D) and one chiral center at position 20 within the alpha-hydroxy lactone ring with (S) configuration (the E-ring). Its planar structure is thought to be one of the most important factors in topoisomerase inhibition.[8][9]
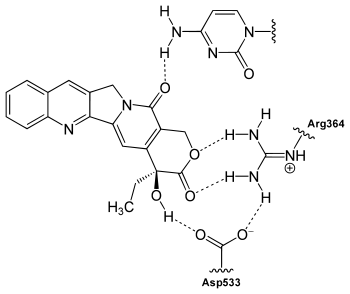
Binding
CPT binds to the topo I and DNA complex (the covalent complex) resulting in a ternary complex, and thereby stabilizing it. This prevents DNA re-ligation and therefore causes DNA damage which results in apoptosis.[10] CPT binds both to the enzyme and DNA with hydrogen bonds. The most important part of the structure is the E-ring which interacts from three different positions with the enzyme. The hydroxyl group in position 20 forms hydrogen bond to the side chain on aspartic acid number 533 (Asp533) in the enzyme. It is critical that the configuration of the chiral carbon is (S) because (R) is inactive. The lactone is bonded with two hydrogen bonds to the amino groups on arginine 364 (Arg364). The D-ring interacts with the +1 cytosine on non-cleaved strand and stabilizes the topo I-DNA covalent complex by forming hydrogen bond. This hydrogen bond is between carbonyl group in position 17 on the D-ring and amino group on the pyrimidine ring of +1 cytosine.[11][12] CPT is selectively cytotoxic to the cells replicating DNA during S phase [13] and its toxicity is primarily a result of conversion of single-strand breaks into double-strand breaks when the replication fork collides with the cleavage complexes formed by DNA and CPT.[14]
Chemistry
The lactone ring in CPT is highly susceptible to hydrolysis. The open ring form is inactive and it must therefore be closed to inhibit topo I. The closed form is favored in acidic condition, as it is in many cancer cells microenvironment. CPT is transported into the cell by passive diffusion. Cellular uptake is favored by lipophilicity, which enhances intracellular accumulation. Lipophilicity makes compounds more stable because of improved lactone partitioning into red blood cells and consequently less hydrolysis of the lactone. CPT has affinity for human serum albumin (HSA), especially the carboxylate form of CPT. Because of that, the equilibrium between the lactone ring and the carboxylate form is driven toward the carboxylate. Reduced drug-HSA interactions could result in improved activity.[11][15]
SAR – Structure-activity relationship
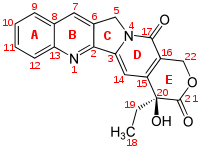
Studies have shown that substitution at position 7, 9, 10 and 11 can have positive effect on CPT activity and physical properties, e.g. potency and metabolic stability. Enlargement of the lactone ring by one CH
2 unit also enhances its abilities, as in homocamptothecin. Substitution at position 12 and 14 leads to inactive derivative.[15]
A- and B-ring modification
Alkyl substitution
Alkyl substitution at position 7 has shown increased cytotoxicity, such as ethyl (C2H5) or chloromethyl (CH2Cl). These groups are able to react with the DNA in the presence of topo I which leads to more tumor activity. It has also been shown that increasing the length of the carbon chain (in position 7) leads to increased lipophilicity and consequently greater potency and stability in human plasma.[11][15] Other 7-modified CPT analogues are silatecans and karenitecins. They are potent inihibitors on topo I and both have alkylsilyl groups in position 7 which make them lipophilic and more stable. Silatecans or 7-silylcampthothecins have shown reduced drug-HSA interactions which contributes to its blood stability and they can also cross the blood brain barrier. DB-67 is a 10-hydroxy derivative and is among the most active silatecans. BNP1350 which belongs to the series of karenitecins exhibits cytotoxic activity and ability to overcome drug resistance. Still another route to make CPT’s lipophilic is to introduce lipophilic substituents, such as iminomethyl or oxyiminomethyl moieties. One of the most potent compounds is the oxyiminomethyl derivative ST1481 that has the advantage to overcome drug resistance caused by transport systems.[15] Basic nitrogen in a carbon chain at position 7 makes the compound more hydrophilic and hence more water-soluble. For example, is a derivate called CKD-602, which is a potent topo I inhibitor and successfully overcomes the poor water solubility and toxicity seen with CPT.[15][16]
Considerably greater activity can be achieved by putting electron-withdrawing groups like amino, nitro, bromo or chloro at position 9 and 10 and hydroxyl group at position 10 or 11. But these compounds are relatively insoluble in aqueous solutions, which causes difficulty in administrations. Methoxy group at both position 10 and 11 simultaneously leads to inactivity.[8][15]
Hexacyclic CPT analogues
Hexacyclic CPT analogues have shown great potency. For example, methylenedioxy or ethylenedioxy group connected between 10 and 11 form a 5 or 6 membered ring which leads to more water-soluble derivates and increased potency. Researches have shown that ethylenedioxy analogues are less potent than methylenedioxy. The reason is the unfavorable steric interactions of ethylenedioxy analogues with the enzyme.[8][15]
Adding amino or chloro group at 9th position or chloromethyl group at 7th position to these 10, 11-methylenedioxy or ethylenedioxy analogues results in compounds with even greater cytotoxicity but weaker solubility in water. To yield 10, 11-methylenedioxy or ethylenedioxy analogues with good water solubility a good way is to introduce a water solubilising substituent at position 7. Lurtotecan meets those requirements; it’s a 10, 11-ethylenedioxy analogue with a 4-methylpiperazino-methylene at position 7 and has shown a great potency in clinical researches.[8]
A ring can also be formed between position 7 and 9, like position 10 and 11. That gives new opportunities to make water-soluble derivatives [5]. These hexacyclic CPT become more active when electron-withdrawing groups are put in position 11 and methyl or amino groups at 10. Exatecan is an example of hexacyclic CPT that has a 6 membered ring over position 7 and 9, and is 10-methyl, 11-fluoro substituted [4]. It is water-soluble and more potent than topotecan.[8][15][17]
C- and D-ring modification
The C- and D-rings have an essential role in the antitumor activity. Replacement in any position results in much less potent compound than parent compound in other cytotoxicity assay.[8]
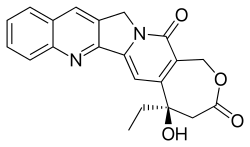
E-ring modifications
The E-ring doesn’t allow many structural changes without losing CPT activity. One possible replacement is changing the hydroxyl group to Cl, F or Br because their polarizability is sufficient to stabilize the enzyme-complex.[15]
Another possible modification is to insert a methylene between hydroxyl and lactone on the E-ring yielding a seven membered β-hydroxylactone group, so-called homocamptothecin (hCPT). The hCPT’s hydroxyl has less inductive effect on the carboxyl group which makes the lactone very reactive. This enhances the interaction of the free hydroxyl group optimally with topo I and the covalent complex that forms in its presence are more stable. The E-ring of hCPT opens more slowly and the opening is irreversible. hCPTs exhibit enhanced human plasma stability because of decreased protein binding and more affinity for red blood cells than CPT.[8][15]
CPT analogues
Since the discovery of CPT many analogues have been synthesized. Below is a schematic view of the CPT analogues that have been mentioned in the text above.
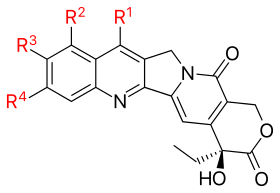 | ||||
| Analogue | R1 | R2 | R3 | R4 |
| Topotecan | —H | 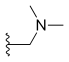 |
—OH | —H |
| Irinotecan (CPT-11) | —H | 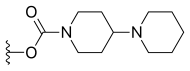 |
—H | |
| Silatecan (DB-67, AR-67) | 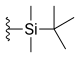 |
—H | —OH | —H |
| Cositecan (BNP-1350) | 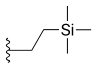 |
—H | —H | —H |
| Exatecan | 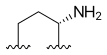 |
—CH3 | —F | |
| Lurtotecan | 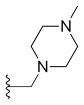 |
—H | 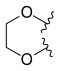 | |
| Gimatecan (ST1481) |  |
—H | —H | —H |
| Belotecan (CKD-602) | 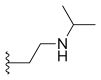 |
—H | —H | —H |
| Rubitecan | —H | 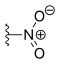 |
—H | —H |
CPT is linked to a cyclodextrin-based polymer to form the investigational anti-cancer drug CRLX101.[18]
References
- . the stem bark of Mappia foetida, a tree native to India, has proved to be another source significant for the isolation of camptothecin.TR Govindachari and N. Viswnathan, Phytochemistry,11(12), 3529-31 (1972). Efferth T, Fu YJ, Zu YG, Schwarz G, Konkimalla VS, Wink M (2007). "Molecular target-guided tumor therapy with natural products derived from traditional Chinese medicine". Current Medicinal Chemistry. 14 (19): 2024–32. doi:10.2174/092986707781368441.
- "Curran Synthesis of Camptothecin". Archived from the original on 2009-09-05.
- "Comins Synthesis of Camptothecin". Archived from the original on 2009-09-05.
- "Rapaport Synthesis of Camptothecin".
- Takimoto CH, Calvo E. "Principles of Oncologic Pharmacotherapy" in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management: A Multidisciplinary Approach. 11 ed. 2008.
- M.E. Wall; M.C.Wani; C.E. Cook; K.H.Palmer; A.I.McPhail; G.A.Sim (1966). "Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminate". J. Am. Chem. Soc. 88 (16): 3888–3890. doi:10.1021/ja00968a057.
- G. Samuelsson (2004). Drugs of Natural Origin: a Textbook of Pharmacognosy (5 ed.). Stokkholm: Swedish pharmaceutical press. ISBN 91-974318-4-2.
- H. Ulukan; P.W. Swaan (2002). "Camptothecins, a review of their chemotherapeutical potential". Drugs (27 ed.). 62 (2): 2039–2057. doi:10.2165/00003495-200262140-00004. PMID 12269849.
- A. J. Lu; Z. S. Zhang; M. Y. Zheng; H. J. Zou; X. M. Luo; H. L. Jiang (2007). "3D-QSAR study of 20 (S)-camptothecin analogs". Acta Pharmacologica Sinica. 28 (2): 307–314. doi:10.1111/j.1745-7254.2007.00477.x.
- "Camptothecin". DrugBank. Retrieved 9 October 2016.
- D. J. Adams; M. L. Wahl; J. L. Flowers; B. Sen; M. Colvin; M. W. Dewhirst; G. Manikumar; M. C. Wani (2005). "Camptothecin analogs with enhanced activity against human breast cancer cells. II. Impact of the tumor pH gradient". Cancer Chemotherapy and Pharmacology. 57 (2): 145–154. doi:10.1007/s00280-005-0008-5. PMID 16001167.
- M. R. Redinbo; L. Stewart; P. Kuhn; J. J. Champoux; W. G. J. Hol (1998). "Crystal structure of human topoisomerase I in covalent and noncovalent complexes with DNA". Science. 279 (5356): 1504–1513. doi:10.1126/science.279.5356.1504. PMID 9488644.
- Del Bino G, Lassota P, Darzynkiewicz Z. (1991) The S-phase cytotoxicity of camptothecin. Exp Cell Res 193:27-35. PMID 1995300
- Y. Pommier; C. Redon; V.A. Rao; J.A. Seiler; O. Sordet; H. Takemura; S. Antony; L. Meng; Z.Liao; G. Kohlhagen (2003). "Repair of and checkpoint response to topoisomerase I-mediated DNA damage". Mutat. Res. 532 (1–2): 173–203. doi:10.1016/j.mrfmmm.2003.08.016. PMID 14643436.
- F. Zunino; S. Dallavalle; D. Laccabue; G. Beretta; L. Merlini; G. Pratesi (2002). "Current status and perspectives in the Development of Camptothecins". Current Pharmaceutical Design (27 ed.). 8 (27): 2505–2520. doi:10.2174/1381612023392801. PMID 12369944.
- M. K. Chung; S. S. Han; J. C. Kim (2006). "Evaluation of the toxic potentials of a new camptothecin anticancer agent CKD-602 on fertility and early embryonic development in rats". Regulatory Toxicology and Pharmacology. 45 (3): 273–281. doi:10.1016/j.yrtph.2006.05.004. PMID 16814440.
- M. Palumbo; C. Sissi; B. Gatto; S. Moro; G. Zagotto (2001). "Quantitation of camptothecin and related compounds". J. Chromatogr. B. 764 (1–2): 121–40. doi:10.1016/S0378-4347(01)00345-0.
- "Cerulean Raises $24M to Progress Clinical Development of Nanopharmaceuticals". 15 Nov 2010.