Acute liver failure
Acute liver failure is the appearance of severe complications rapidly after the first signs (such as jaundice) of liver disease, and indicates that the liver has sustained severe damage (loss of function of 80–90% of liver cells). The complications are hepatic encephalopathy and impaired protein synthesis (as measured by the levels of serum albumin and the prothrombin time in the blood). The 1993 classification defines hyperacute as within 1 week, acute as 8–28 days, and subacute as 4–12 weeks.[1] It reflects the fact that the pace of disease evolution strongly influences prognosis. Underlying cause is the other significant determinant of outcome.[2]
| Acute liver failure | |
|---|---|
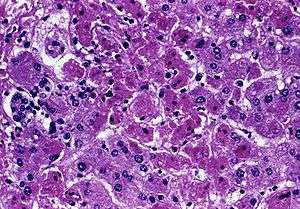 | |
| Acute liver failure (with hepatocellular necrosis and sinusoidal bleeding) from Marburg virus, a rare cause | |
| Specialty | Gastroenterology, hepatology, intensive care medicine |
| Symptoms | Jaundice, excessive bleeding, altered state of consciousness, hyperdynamic circulation, hepatocellular necrosis, low blood pressure |
| Complications | Hepatic encephalopathy, kidney failure, thrombocytopenia, hyponatraemia |
| Diagnostic method | Prothrombin time measurement, complete blood count |
| Treatment | Liver transplant |
Signs and symptoms
The main features of acute liver failure are rapid-onset jaundice, weakness, and eventually, changes in mental status that can begin as mild confusion but progress to coma.
Encephalopathy and cerebral edema
In ALF, hepatic encephalopathy leads to cerebral edema, coma, brain herniation, and eventually death. Detection of encephalopathy is central to the diagnosis of ALF. It may vary from subtle deficit in higher brain function (e.g. mood, concentration in grade I) to deep coma (grade IV). Patients presenting as acute and hyperacute liver failure are at greater risk of developing cerebral oedema and grade IV encephalopathy. The pathogenesis remains unclear, but is likely to be a consequence of several phenomena. There is a buildup of toxic substances like ammonia, mercaptan, benzodiazepines, serotonin and tryptophan in the brain. This affects neurotransmitter level and neuroreceptor activation. Autoregulation of cerebral blood flow is impaired, and is associated with anaerobic glycolysis and oxidative stress. Neuronal cell astrocytes are susceptible to these changes, and they swell up, resulting in increased intracranial pressure. Inflammatory mediators also play important role.[2][3][4]
Unfortunately, signs of elevated intracranial pressure, such as papilledema and loss of pupillary reflexes, are not reliable, and occur late in the disease process. CT imaging of the brain is also unhelpful in detecting early cerebral oedema, but is often performed to rule out intra-cerebral bleeding. Invasive intracranial pressure monitoring via subdural route is often recommended; however, the risk of complications must be weighed against the possible benefit (1% fatal haemorrhage).[5] The aim is to maintain intracranial pressures below 25 mm Hg, and cerebral perfusion pressures above 50 mm Hg.[2]
Coagulopathy
Coagulopathy is another cardinal feature of ALF. The liver has the central role in the synthesis of almost all coagulation factors and some inhibitors of coagulation and fibrinolysis. Hepatocellular necrosis leads to impaired synthesis of many coagulation factors and their inhibitors. The former produces a prolongation in prothrombin time which is widely used to monitor the severity of hepatic injury. There is significant platelet dysfunction (with both quantitative and qualitative platelet defects). Progressive thrombocytopenia with the loss of larger and more active platelets is almost universal. Thrombocytopenia with or without DIC increases risk of intracerebral bleeding.[6]
Kidney failure
Kidney failure is common, present in more than 50% of ALF patients, either due to original insult such as paracetamol resulting in acute tubular necrosis or from hyperdynamic circulation leading to hepatorenal syndrome or functional kidney failure. Because of impaired production of urea, blood urea does not represent the degree of kidney impairment.
Inflammation and infection
About 60% of all ALF patients fulfil the criteria for systemic inflammatory syndrome irrespective of presence or absence of infection.[7] This often contributes towards multi organ failure. Impaired host defence mechanism, due to impaired opsonization, chemotaxis and intracellular killing, substantially increases risk of sepsis. Bacterial sepsis mostly due to gram positive organisms and fungal sepsis are observed in up to 80% and 30% patients, respectively.[6]
Metabolic derangements
Hyponatraemia is an almost universal finding due to water retention and a shift in intracellular sodium transport from inhibition of Na/K ATPase. Hypoglycaemia (due to depleted hepatic glycogen store and hyperinsulinaemia), hypokalaemia, hypophosphataemia and metabolic alkalosis are often present, independent of renal function. Lactic acidosis occurs predominantly in paracetomol (also known as acetaminophen) overdose.
Haemodynamic and cardio-respiratory compromise
Hyperdynamic circulation, with peripheral vasodilatation from low systemic vascular resistance, leads to hypotension. There is a compensatory increase in cardiac output. Adrenal insufficiency has been documented in 60% of ALF cases, and is likely to contribute in haemodynamic compromise.[8] There is also abnormal oxygen transport and utilization. Although delivery of oxygen to the tissues is adequate, there is a decrease in tissue oxygen uptake, resulting in tissue hypoxia and lactic acidosis.[9]
Pulmonary complications occur in up to 50% of patients.[10] Severe lung injury and hypoxemia result in high mortality. Most cases of severe lung injury are due to ARDS, with or without sepsis. Pulmonary haemorrhage, pleural effusions, atelectasis, and intrapulmonary shunts also contribute to respiratory difficulty.
Late pregnancy
In late pregnancy liver function decreases significantly, which can be easily monitored by blood tests. Early clinical manifestations of ALF in late pregnancy include hypodynamia, decrease in appetite, dark amber urine, deep jaundice, nausea, vomiting, and abdominal distention.[11] Among patients whose deaths were attributed to ALF in late pregnancy, the majority had experienced vaginal deliveries.[12]
Causes
Common causes for acute liver failure are paracetamol (acetaminophen) overdose, idiosyncratic reaction to medication (e.g. tetracycline, troglitazone), excessive alcohol consumption (severe alcoholic hepatitis), viral hepatitis (hepatitis A or B — it is extremely uncommon in hepatitis C), acute fatty liver of pregnancy, and idiopathic (without an obvious cause). Reye syndrome is acute liver failure in a child with a viral infection (e.g. chickenpox); it appears that aspirin use may play a significant role. Wilson's disease (hereditary copper accumulation) may infrequently present with acute liver failure. Acute liver failure also results from poisoning by the death cap mushroom (Amanita phalloides) as well as other amatoxin-producing fungus species. Certain strains of Bacillus cereus - a common species of bacterium implicated as a frequent cause of food poisoning - can cause fulminant liver failure through the production of cereulide,[13] a toxin which destroys the mitochondria in affected hepatocytes, resulting in cell death. While most instances of B. cereus infection are resolved by the body's immune system and do not affect the liver, severe cases resulting in liver damage can be fatal[14][15][16][17][18] without immediate treatment or liver transplantation.
Pathophysiology
In the majority of acute liver failure (ALF) there is widespread hepatocellular necrosis beginning in the centrizonal distribution and progressing towards portal tracts. The degree of parenchymal inflammation is variable and is proportional to duration of disease.[6][19]
Zone 1 (periportal) occurs in phosphorus poisoning or eclampsia. Zone 2 (mid-zonal), although rare, is seen in yellow fever. Zone 3 (centrilobular) occurs with ischemic injury, toxic effects, carbon tetrachloride exposure, or chloroform ingestion. In acute acetaminophen overdose, toxification occurs, mostly in Zone III which has the highest level of P450 micro-enzymes. That fact along with Zone III's decreased oxygen level helps to explain why it is preferentially one of the initial sites of damage.
Diagnosis
All patients with clinical or laboratory evidence of moderate to severe acute hepatitis should have an immediate measurement of prothrombin time and careful evaluation of mental status. If the prothrombin time is prolonged by ≈ 4–6 seconds or more (INR ≥ 1.5), and there is any evidence of altered sensorium, the diagnosis of ALF should be strongly suspected, and hospital admission is mandatory.[20] Initial laboratory examination must be extensive in order to evaluate both the etiology and severity.
- Initial laboratory analysis[20]
- Prothrombin time/INR
- Complete blood count
- Chemistries
- Liver function test: AST, ALT, alkaline phosphatase, GGT, total bilirubin, albumin
- Creatinine, urea/blood urea nitrogen, sodium, potassium, chloride, bicarbonate, calcium, magnesium, phosphate
- Glucose
- Amylase and lipase
- Arterial blood gas, lactate
- Blood type and screen
- Paracetamol (acetaminophen) level, toxicology screen
- Viral hepatitis serologies: anti-HAV IgM, HBSAg, anti-HBc IgM, anti-HCV
- Autoimmune markers: ANA, ASMA, LKMA, immunoglobulin levels
- Ceruloplasmin level (when Wilson's disease suspected)
- Pregnancy test (females)
- Ammonia (arterial if possible)
- HIV status (has implication for transplantation)
History taking should include a careful review of possible exposures to viral infection and drugs or other toxins. From history and clinical examination, the possibility of underlying chronic disease should be ruled out as it may require different management.
A liver biopsy done via the transjugular route because of coagulopathy is not usually necessary, other than in occasional malignancies. As the evaluation continues, several important decisions have to be made; such as whether to admit the patient to an ICU, or whether to transfer the patient to a transplant facility. Consultation with the transplant center as early as possible is critical due to the possibility of rapid progression of ALF.
Definition
Acute liver failure is defined as "the rapid development of hepatocellular dysfunction, specifically coagulopathy and mental status changes (encephalopathy) in a patient without known prior liver disease".[21]page 1557
The diagnosis of acute liver failure is based on physical exam, laboratory findings, patient history, and past medical history to establish mental status changes, coagulopathy, rapidity of onset, and absence of known prior liver disease respectively.[21]page 1557
The exact definition of "rapid" is somewhat questionable, and different sub-divisions exist which are based on the time from onset of first hepatic symptoms to onset of encephalopathy. One scheme defines "acute hepatic failure" as the development of encephalopathy within 26 weeks of the onset of any hepatic symptoms. This is sub-divided into "fulminant hepatic failure", which requires onset of encephalopathy within 8 weeks, and "subfulminant", which describes onset of encephalopathy after 8 weeks but before 26 weeks.[22] Another scheme defines "hyperacute" as onset within 7 days, "acute" as onset between 7 and 28 days, and "subacute" as onset between 28 days and 24 weeks.[21]page 1557
Treatment
| King's College Hospital criteria for liver transplantation in acute liver failure[23] |
| Patients with paracetamol toxicity pH < 7.3 or |
| Other patients Prothrombin time > 100 seconds or
|
General concerns
Because ALF often involves the rapid deterioration of mental status and the potential for multiorgan failure, patients should be managed in the intensive care unit.[24] For patients not at a transplant center, the possibility of rapid progression of ALF makes early consultation with a transplant facility critical. Accordingly, plans for transfer to a transplant center should begin in patients with any abnormal mentation. Early institution of antidotes or specific therapy may prevent the need for liver transplantation and reduce the likelihood of poor outcome. Measures appropriate for specific causes of ALF are described in detail later in this chapter.[25]
Neurologic complications
Patients with grade I–II encephalopathy should be transferred to a liver transplant facility and listed for transplantation. Consider a brain computed tomography (CT) scan to rule out other causes of altered or impaired mental status. Stimulation and overhydration can cause elevations in intracranial pressure (ICP) and should be avoided. Unmanageable agitation may be treated with short-acting benzodiazepines in small doses. Lactulose can be considered at this stage. A preliminary report from the ALFSG on 117 patients suggests that use of lactulose in the first 7 days after diagnosis is associated with a small increase in survival time, but with no difference in severity of encephalopathy or in the overall outcome. For patients who progress to grade III–IV encephalopathy, intubation for airway protection is generally required. Many centers use propofol for sedation because it may reduce cerebral blood. The head of the bed should be elevated to 30 degrees, and electrolytes, blood gasses, glucose, and neurologic status monitored frequently.[26][27]
Cardiovascular complications
Increased cardiac output and low systemic vascular resistance are characteristic of ALF. Pulmonary artery catheterization should be considered. Hypotension should be treated preferentially with fluids, but systemic vasopressor support with agents such as epinephrine, norepinephrine, or dopamine should be used if fluid replacement fails to maintain mean arterial pressure of 50–60 mm Hg. Vasoconstrictive agents (especially vasopressin) should be avoided.[28]
Pulmonary complications
Pulmonary edema and pulmonary infections are commonly seen in patients with ALF. Mechanical ventilation may be required. However, positive end-expiratory pressure can worsen cerebral edema.[29]
Coagulopathy and gastrointestinal bleeding
Impaired liver synthesis of clotting factors, low-grade fibrinolysis, and intravascular coagulation are typical of ALF. Thrombocytopenia is common and may also be dysfunctional. Replacement therapy is recommended only in the setting of bleeding or prior to an invasive procedure. Vitamin K can be given to treat an abnormal prothrombin time, regardless of whether there is poor nutritional status. Administration of recombinant factor VIIa has shown promise; however, this treatment approach requires further study. The use of gastrointestinal hemorrhage prophylaxis with a histamine-2 (H2) blocker, proton pump inhibitor, or sucralfate is recommended.[30]
Nutrition, electrolytes, and metabolic derangements
In patients with grade I or II encephalopathy, enteral feeding should be initiated early. Parenteral nutrition should be used only if enteral feeding is contraindicated as it increases the risk of infection. Severe restriction of protein is not beneficial; 60 g/day of protein is generally reasonable. Fluid replacement with colloid (e.g. albumin) is preferred rather than crystalloid (e.g. saline); all solutions should contain dextrose to maintain euglycemia. Multiple electrolyte abnormalities are common in ALF. Correction of hypokalemia is essential as hypokalemia increases the kidneys' ammonia production, potentially exacerbating encephalopathy. Hypophosphatemia is especially common in patients with acetaminophen-induced ALF and in those with intact renal function. Hypoglycemia occurs in many patients with ALF and is often due to depletion of hepatic glycogen stores and impaired gluconeogenesis. Plasma glucose concentration should be monitored and hypertonic glucose administered as needed.[31]
Infection
Bacterial and fungal infections are common in ALF, with one study demonstrating culture-proven infection in 80% of ALF patients. Defective cellular and humoral immunity as well as presence of indwelling catheters, coma, broad-spectrum antibiotics, and medications that suppress immunity all predispose to infection. Localizing symptoms of infection such as fever and sputum production are frequently absent and the only clues to an underlying infectious process may be worsening of encephalopathy or renal function. There must be a low threshold for obtaining frequent cultures (blood, urine, and sputum), chest radiographs, and paracentesis. Bacteria that enter through the skin, such as streptococci and staphylococci, tend to predominate. Aggressive surveillance is essential as prophylactic antibiotics have shown little benefit. Fungal infections, particularly in the setting of broad-spectrum antibiotics, are also common, and disseminated fungemia is a poor prognostic sign.[32]
Liver transplantation
The advent of transplantation has changed survival from as low as 15% in the pretransplant era to more than 60% today. Liver transplantation is indicated for many patients with ALF, and survival rates of 56–90% can be achieved. In addition to transplantation, better critical care and the trend toward more benign causes, such as acetaminophen, all contribute to improved survival rates. Spontaneous survival is now around 40%. The application of transplantation among patients with ALF remains low, suggesting that the full potential of this modality may not be realized. Timely availability of an allograft is one of the major factors determining transplant outcomes. In the largest U.S. study, only 29% of patients received a liver graft, while 10% of the overall group (one fourth of patients listed for transplantation) died on the waiting list. Other series have reported death rates of those listed for transplant as high as 40%. In the ALFSG, the transplantation rate was higher in the groups with lower short-term spontaneous survival, making overall survival similar in all groups: acetaminophen, 73%; drug induced, 70%; indeterminate group, 64%; and other causes, 61%. Causes of death for the 101 patients who died within the 3-week period included cerebral edema, multiorgan failure, sepsis, cardiac arrhythmia or arrest and respiratory failure. The median time to death after admission was 5 days.[32]
Acetylcysteine
Intravenous N-acetylcysteine has been found to be beneficial in both acetaminophen toxicity and non-acetaminophen-related acute liver failure.[33]
Prognosis
Historically mortality has been high, being in excess of 80%.[34] In recent years the advent of liver transplantation and multidisciplinary intensive care support have improved survival significantly. At present overall short-term survival with transplant is more than 65%.[35]
Several prognostic scoring systems have been devised to predict mortality and to identify who will require an early liver transplant. These include King's College Hospital criteria, MELD score, APACHE II, and Clichy criteria.
Terminology
To date, no universally accepted nomenclature has been adopted. Trey and Davidson introduced the phrase fulminant hepatic failure in 1970, which they described as a "... potentially reversible condition, the consequence of severe liver injury, with an onset of encephalopathy within 8 weeks of the appearance of the first symptoms and in the absence of pre-existing liver disease".[36] Later, it was suggested that the term fulminant should be confined to patients who develop jaundice to encephalopathy within 2 weeks. Phrases subfulminant hepatic failure and late onset' hepatic failure were coined for onset between 2 weeks to 3 months and for 8 weeks to 24 weeks, respectively.[37][38] The umbrella phrase of acute liver failure was proposed by King's College group, which has been adopted in this article. Paradoxically, in this classification, the best prognosis is in the hyperacute group.[39]
References
- O'Grady JG, Schalm SW, Williams R (1993). "Acute liver failure: redefining the syndromes". Lancet. 342 (8866): 273–5. doi:10.1016/0140-6736(93)91818-7. PMID 8101303.
- O'Grady JG (2005). "Acute liver failure". Postgraduate Medical Journal. 81 (953): 148–54. doi:10.1136/pgmj.2004.026005. PMC 1743234. PMID 15749789.
- Hazell, Alan S.; Butterworth, Roger F. (1999). "Hepatic encephalopathy: An update of pathophysiologic mechanisms". Proc. Soc. Exp. Biol. Med. 222 (2): 99–112. doi:10.1046/j.1525-1373.1999.d01-120.x. PMID 10564534.
- Larsen FS, Wendon J (2002). "Brain edema in liver failure: basic physiologic principles and management". Liver Transpl. 8 (11): 983–9. doi:10.1053/jlts.2002.35779. PMID 12424710.
- Armstrong IR, Pollok A, Lee A (1993). "Complications of intracranial pressure monitoring in fulminant hepatic failure". Lancet. 341 (8846): 690–1. doi:10.1016/0140-6736(93)90458-S. PMID 8095592.
- Gimson AE (1996). "Fulminant and late onset hepatic failure". British Journal of Anaesthesia. 77 (1): 90–8. doi:10.1093/bja/77.1.90. PMID 8703634.
- Schmidt LE, Larsen FS (2006). "Prognostic implications of hyperlactatemia, multiple organ failure, and systemic inflammatory response syndrome in patients with acetaminophen-induced acute liver failure". Crit. Care Med. 34 (2): 337–43. doi:10.1097/01.CCM.0000194724.70031.B6. PMID 16424712.
- Harry R, Auzinger G, Wendon J (2002). "The clinical importance of adrenal insufficiency in acute hepatic dysfunction". Hepatology. 36 (2): 395–402. doi:10.1053/jhep.2002.34514. PMID 12143048.
- Bihari D, Gimson AE, Waterson M, Williams R (1985). "Tissue hypoxia during fulminant hepatic failure". Crit. Care Med. 13 (12): 1034–9. doi:10.1097/00003246-198512000-00010. PMID 3933911.
- Trewby PN, Warren R, Contini S, et al. (1978). "Incidence and pathophysiology of pulmonary edema in fulminant hepatic failure". Gastroenterology. 74 (5 Pt 1): 859–65. doi:10.1016/0016-5085(78)90142-7. PMID 346431.
- Li XM, Ma L, Yang YB, Shi ZJ, Zhou SS (2005). "Clinical characteristics of fulminant hepatitis in pregnancy". World J Gastroenterol. 11 (29): 4600–3. doi:10.3748/wjg.v11.i29.4600. PMC 4398717. PMID 16052697.
- Li XM, Ma L, Yang YB, Shi ZJ, Zhou SS (2005). "Prognostic factors of fulminant hepatitis in pregnancy". Chin Med J (Engl). 118 (20): 1754–7. PMID 16313765.
- Peltola; et al. (2004). "News on cereulide, the emetic toxin of Bacillus Cereus".
- Takabe F, Oya M (1976). "An autopsy case of food poisoning associated with Bacillus cereus". Forensic Science. 7 (2): 97–101. doi:10.1016/0300-9432(76)90024-8. PMID 823082.
- Mahler H; et al. (1997). "Fulminant liver failure in association with the emetic toxin of Bacillus cereus". N Engl J Med. 336 (16): 1142–1148. doi:10.1056/NEJM199704173361604. PMID 9099658.
- Dierick K; et al. (2005). "Fatal family outbreak of Bacillus cereus-associated food poisoning". J Clin Microbiol. 43 (8): 4277–4279. doi:10.1128/JCM.43.8.4277-4279.2005. PMC 1233987. PMID 16082000.
- Shiota, M; et al. (2010). "Rapid Detoxification of Cereulide in Bacillus cereus Food Poisoning". Pediatrics. 125 (4): e951–e955. doi:10.1542/peds.2009-2319. PMID 20194285.
- Naranjo, M; et al. (2011). "Sudden Death of a Young Adult Associated with Bacillus cereus Food Poisoning". J Clin Microbiol. 49 (12): 4379–4381. doi:10.1128/JCM.05129-11. PMC 3232990. PMID 22012017.
- Boyer JL, Klatskin G (1970). "Pattern of necrosis in acute viral hepatitis. Prognostic value of bridging (subacute hepatic necrosis)". N. Engl. J. Med. 283 (20): 1063–71. doi:10.1056/NEJM197011122832001. PMID 4319402.
- Polson J, Lee WM (2005). "AASLD position paper: the management of acute liver failure". Hepatology. 41 (5): 1179–97. doi:10.1002/hep.20703. PMID 15841455.
- Sleisenger, edited by Mark Feldman, Lawrence S. Friedman, Lawrence J. Brandt; consulting editor, Marvin H. (2009). Sleisenger & Fordtran's gastrointestinal and liver disease pathophysiology, diagnosis, management (PDF) (9th ed.). St. Louis, Mo.: MD Consult. ISBN 978-1-4160-6189-2.CS1 maint: extra text: authors list (link)
- Sood, Gagan K. "Acute Liver Failure". Mescape. Retrieved 14 December 2011.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R (1989). "Early indicators of prognosis in fulminant hepatic failure". Gastroenterology. 97 (2): 439–45. doi:10.1016/0016-5085(89)90081-4. PMID 2490426.
- Jalan, R (2005). "Acute liver failure: current management and future prospects". Journal of Hepatology. 42 Suppl (1): S115–23. doi:10.1016/j.jhep.2004.11.010. PMID 15777566.
- Polson, J; Lee, WM; American Association for the Study of Liver, Disease (May 2005). "AASLD position paper: the management of acute liver failure". Hepatology. 41 (5): 1179–97. doi:10.1002/hep.20703. PMID 15841455.
- Jalan, R (August 2003). "Intracranial hypertension in acute liver failure: pathophysiological basis of rational management". Seminars in Liver Disease. 23 (3): 271–82. doi:10.1055/s-2003-42645. PMID 14523680.
- Jalan, R; Olde Damink, SW; Deutz, NE; Davies, NA; Garden, OJ; Madhavan, KK; Hayes, PC; Lee, A (27 June 2003). "Moderate hypothermia prevents cerebral hyperemia and increase in intracranial pressure in patients undergoing liver transplantation for acute liver failure". Transplantation. 75 (12): 2034–9. doi:10.1097/01.tp.0000066240.42113.ff. PMID 12829907.
- Murphy, N; Auzinger, G; Bernel, W; Wendon, J (February 2004). "The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure". Hepatology. 39 (2): 464–70. doi:10.1002/hep.20056. PMID 14767999.
- Wijdicks, EF; Nyberg, SL (June 2002). "Propofol to control intracranial pressure in fulminant hepatic failure". Transplantation Proceedings. 34 (4): 1220–2. doi:10.1016/s0041-1345(02)02804-x. PMID 12072321.
- Shami, VM; Caldwell, SH; Hespenheide, EE; Arseneau, KO; Bickston, SJ; Macik, BG (February 2003). "Recombinant activated factor VII for coagulopathy in fulminant hepatic failure compared with conventional therapy". Liver Transplantation. 9 (2): 138–43. doi:10.1053/jlts.2003.50017. PMID 12548507.
- Brown RS, Jr; Russo, MW; Lai, M; Shiffman, ML; Richardson, MC; Everhart, JE; Hoofnagle, JH (27 February 2003). "A survey of liver transplantation from living adult donors in the United States". The New England Journal of Medicine. 348 (9): 818–25. doi:10.1056/nejmsa021345. PMID 12606737.
- Farmer, DG; Anselmo, DM; Ghobrial, RM; Yersiz, H; McDiarmid, SV; Cao, C; Weaver, M; Figueroa, J; Khan, K; Vargas, J; Saab, S; Han, S; Durazo, F; Goldstein, L; Holt, C; Busuttil, RW (May 2003). "Liver transplantation for fulminant hepatic failure: experience with more than 200 patients over a 17-year period". Annals of Surgery. 237 (5): 666–75, discussion 675–6. doi:10.1097/01.sla.0000064365.54197.9e. PMC 1514517. PMID 12724633.
- Lee WM, Hynan LS, Rossaro L, et al. (September 2009). "Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure". Gastroenterology. 137 (3): 856–64, 864.e1. doi:10.1053/j.gastro.2009.06.006. PMC 3189485. PMID 19524577.
- Rakela J, Lange SM, Ludwig J, Baldus WP (1985). "Fulminant hepatitis: Mayo Clinic experience with 34 cases". Mayo Clin. Proc. 60 (5): 289–92. doi:10.1016/s0025-6196(12)60534-5. PMID 3921780.
- Ostapowicz G, Fontana RJ, Schiødt FV, et al. (2002). "Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States". Ann. Intern. Med. 137 (12): 947–54. doi:10.7326/0003-4819-137-12-200212170-00007. PMID 12484709.
- Trey C, Davidson CS (1970). "The management of fulminant hepatic failure". Progress in Liver Diseases. 3: 282–98. PMID 4908702.
- Bernuau J, Goudeau A, Poynard T, et al. (1986). "Multivariate analysis of prognostic factors in fulminant hepatitis B". Hepatology. 6 (4): 648–51. doi:10.1002/hep.1840060417. PMID 3732998.
- Gimson AE, O'Grady J, Ede RJ, Portmann B, Williams R (1986). "Late onset hepatic failure: clinical, serological and histological features". Hepatology. 6 (2): 288–94. doi:10.1002/hep.1840060222. PMID 3082735.
- Sass DA, Shakil AO (2005). "Fulminant hepatic failure". Liver Transpl. 11 (6): 594–605. doi:10.1002/lt.20435. PMID 15915484.
External links
| Classification | |
|---|---|
| External resources |